Retour à l' index général du cours de chimie on line.
I.1.
Propriétés physiques de l'eau.
I.2. Propriétés
physico-chimiques de l'eau: les ions de l'eau pure. L'autoprotolyse de
l'eau.
I.3. Définition
du pH.
II.1. L'évolution des notions au cours de l'Histoire des Sciences.
II.1.a. La théorie d'ARRHENIUS.II.2. La théorie de BRONSTED développée.
II.1.b. La théorie de BRONSTED.
II.1.c. La théorie de LEWIS.
II.2.a. Les couples acido-basiques de l'eau.
II.2.a.a. L'eau est une base de BRONSTED.II.2.b. Les autres couples acido-basiques dans l'eau.
II.2.a.b. L'eau est un acide de BRONSTED.
II.2.a.g. L'eau est un solvant amphotère.
II.3.
Les pH des solutions aqueuses simples.
II.3.a. Les monoacides forts.
II.3.b. Les monobases fortes.
II.3.c. Les monoacides faibles.
II.3.d. Les monobases faibles.
II.3.e. Les polyacides faibles.
II.3.f. Les polybases faibles.
II.3.g. Les acides aminés
III.
Les sels et les hydrogénosels.
III.1.
Définition de la réaction de salification.
III.2.
Calcul du pH d'un sel d'acide fort et de base forte.
III.3.
Calcul du pH d'un sel d'acide faible et de base forte.
III.4.
Calcul du pH d'un sel d'acide fort et de base faible.
III.5.
Calcul du pH d'un sel d'acide faible et de base faible.
III.6.
Calcul du pH d'un hydrogénosel.
IV. Les courbes de dosages acido-basiques.
IV.1.
Dosage d'un acide fort par une base forte.
IV.2.
Dosage d'un acide faible par une base forte.
IV.3.
Dosage d'une base faible par un acide fort.
IV.4.
Dosage d'un acide faible par une base faible.
IV.5.
Dosage d'un polyacide faible par une base forte.
IV.5.a. Conditions sur les valeurs de deux pKa consécutifs afin de doser séparément deux acidités.IV.5.b.a. Dosage d'un diacide faible: cas où D pKa est supérieur à 4, l'acide sulfureux.
IV.5.b.b. Dosage d'un diacide faible: cas où D pKa est inférieur à 4, l'acide tartrique.
IV.5.b.g. Dosage d'un diacide idéal: le conte de fées.
IV.5.b.d. Dosage d'un triacide faible: cas où D pKa est supérieur à 4, l'acide phosphorique.
IV.5.b.e. Dosage d'un triacide faible: cas où D pKa est inférieur à 4, l'acide citrique.
IV.6.
Dosage d'une polybase faible, le carbonate de sodium.
IV.7.
Dosage d'un mélange de soude et de carbonate de sodium, la soude
carbonatée.
IV.8.
Dosage d'un mélange d'acide chlorhydrique et d'acide phosphorique.
V.1.
Mise en évidence de l'effet tampon.
V.2.
Mesure du pouvoir tampon b d'une solution.
V.3.
Réalisation de solutions tampons.
VI. Les indicateurs
colorés.
VII.
Les électrodes utilisées en pH-métrie.
VIII. Techniques
complémentaires à la pH-métrie: la conductimétrie.
IX.
Exercices.
X.
Corrigés des exercices.
X.1.
Première partie. (jusqu'à l'exercice
IV.9).
X.2.
Seconde partie. (à partir de l'exercice
IV.10).
XI. Conclusion générale.
Bibliographie générale (non exhaustive):
Retour au plan du cours sur les pH des solutions aqueuses.
Je me permets aussi d'ajouter mon commentaire sur divers ouvrages allant de" très juste" à "très complet".
1. Collection Chimie, Hachette Prépa. DURUPTHY et coll. Couleur "bleue" pour les tomes de Maths Sup. et "verte" pour ceux de Maths Spé. TRES BIEN.....
2. Chimie générale. Peter ATKINS. Intereditions. Paris. A regarder.
3. Cours de chimie physique. Paul ARNAUD. Dunod. A regarder. Bien en chimie physique mais peu complet sur cette question des pH...
4. Solutions aqueuses, acides et bases. Chimie des solutions. LE FLOC'H et LACOURBAS. Dunod. TRES BIEN, QUOIQUE MEME TROP COMPLET.
5. Précis de chimie. Tome : chimie des solutions. QUEYREL et MESPLEDE. BREAL éditeur. BIEN.Un bon résumé de cours aussi....
6. Chimie générale. OUHAES et DEVALLEZ. A regarder.
7. Chimie. B. MAHAN. A regarder.
8. Encyclopaedia universalis. Chapitre "Acides et bases". A lire. Très complète pour la chimie en solvant non aqueux.
9. 144 expériences de chimie générale. M. DEFRANCESCHI. Ellipses. De bonnes manips en solvant eau ou solvant non aqueux.
10. 70 expériences de chimie générale. F. RAFFLEGEAU et BRENON. Dunod. De très bonnes manips.
Retour au plan du cours sur les pH des solutions aqueuses.
Retour au plan du cours sur les pH des solutions aqueuses.
Retour au plan du cours sur les pH des solutions aqueuses.
Longueur de liaison O-H, environ
100 pm.
Angle HOH environ 105°. L'eau
se décrit dans la théorie V.S.E.P.R. de GILLESPIE comme étant
un composé de type "AX2E2". Il y a une répulsion
entre les deux doublets non liants de la molécule, répulsion
qui crée comme un effet de cisaillement, d'où la valeur de
105°, différente de 109° 28', angle caractéristique
de la molécule de méthane "AX4".
L'eau est une molécule polaire,
du fait de la différence d'électronégativité
assez forte entre l'hydrogène et l'oxygène.
Electronégativités ![]() de H et O, dansl'échelle d'électronégativité
de
Linus PAULING, Prix
NOBEL de Chimie en 1954, Américain
: 2.1 et 3.5 respectivement.
de H et O, dansl'échelle d'électronégativité
de
Linus PAULING, Prix
NOBEL de Chimie en 1954, Américain
: 2.1 et 3.5 respectivement.
L'eau constitue un dipôle
électrique permanent. L'eau possède un moment dipolaire µ
(voir cours de physique, électrostatique).
Centre
de gravité des charges -
Centre de gravité des charges +
µ= q.l
L'unité SI du moment dipolaire est le coulomb mètre, symbole Cm (pour ne pas confondre avec le millicoulomb, noté mC, qui n'a rien à voir) . Toutefois, dans la pratique on utilise une unité usuelle, appelée le debye, symbole D, car les valeurs de µ en Cm sont très "petites", de l'ordre de 10-29 ou 10-30. James DEBYE était un chimiste néerlandais, Prix NOBEL de Chimie en 1936.
1D = 3.30.10-30Cm
Le moment dipolaire µ de l'eau vaut 1.85 D.
Remarque: l'orientation du
vecteur µ est, ou a été longtemps, sujette à
"controverse" entre chimistes et physiciens!
Selon l'ouvrage que vous consulterez,
selon son "âge", vous pourrez rencontrer µ orienté
du moins vers le plus, ou le contraire! La convention "actuelle" est d'orienter
le vecteur µ du moins vers le plus, un peu comme en électricité
dans le sens des potentiels croissants.
Qu'importe, pourrait-on presque
dire, ce qui compte c'est de bien comprendre que µ traduit une dissymétrie
permanente dans la répartition des charges électriques dans
une molécule.
REM 1: des molécules telles que H2, Cl2,, O2 ,ou CO2, linéaire, ne possèdent pas de moment dipolaire µ permanent, c'est ce qui expliquera leur faible solubilité dans le solvant H2O qui, lui, rappelons-le, en possède: les semblables dissolvent les semblables!
REM 2: l'existence du moment dipolaire µ de l'eau est mise à profit dans les fours à micro-ondes. Le changement, lors de l'alternance du rayonnement électromagnétique émis par la source du four, provoque des frottements entre les molécules d'eau, ce qui se traduit par un échauffement des aliments.
REM 3: du fait de l'existence de µ pour l'eau, du fait aussi de sa valeur "élevée", les propriétés physiques de l'eau sont très "particulières" vis à vis d'éléments situés dans la même colonne du tableau périodique , la colonne des "chalcogènes". C'est ce qu'on appelle "l'anomalie" du point d'ébullition de l'eau. Les exemples expérimentaux sont les suivants:
Eb760 H2S liq = -60.0 °C
Eb760 H2Se liq = - 42.0 °C
Eb760 H2Te liq = -2.0 °C

Pourquoi cela?
Il s'établit dans l'eau
liquide des liaisons intermoléculaires de type "attraction-répulsion"
dipôle-dipôle qu'on appelle "interactions de van der WAALS"
(Prix
NOBEL
de
Physique,
je dis bien Physique,
en 1910, Néerlandais) et, dans
le cas des éléments de la deuxième période
du tableau périodique, dont justement fait partie l'oxygène,
ces liaisons de van der WAALS portent un nom particulier:
les liaisons " hydrogène". C'est leur valeur élevée
du point de vue énergétique (environ 50kJ.mol-1)
qui est la cause de cette valeur "anormalement" élevée du
point d'ébullition de l'eau. Pour passer de l'eau liquide à
l'eau vapeur il faut donc vaincre ces interactions intermoléculaires
et donc chauffer de façon "importante", d'où la valeur de
Eb760 H2O liq.
Ces forces s'exercent à
courte distance: elles varient en ![]() ,
alors que les forces coulombiennes varient en
,
alors que les forces coulombiennes varient en ![]() .
.
Ces interactions "hydrogène"
sont très importantes, très répandues aussi: on se
remémorera que ce sont elles qui donnent, par exemple, sa forme
spatiale à la double hélice de l'ADN.
Insérer schéma des interactions intermoléculaires
entre molécules d'eau.
On rappellera encore que ces interactions intermoléculaires de van der WAALS et hydrogène sont énergétiquement beaucoup plus faibles que n'est une liaison de covalence dont l'ordre de grandeur tourne autour de 400 à 500 kJ.mol-1.
Les conséquences des interactions
de van der WAALS pour l'eau sont multiples:
1. Solubilisation élevée
des espèces polaires. "Les semblables dissolvent les semblables."
2. Ionisation de certaines
molécules: la dissolution de HCl dans l'eau conduit à la
formation d'acide chlorhydrique, c'est à dire, de façon simplifiée,
à H3O+ et Cl -.
3. Solvatation des espèces
formées.
On donne ici l'exemple de l'aqua
complexe de l'ion Fe(III), l'ion hexaquofer(III).

4. Dispersion des ions formés
dans la solution.
L'eau a une constante diélectrique
relative er égale
à 80. On dit que l'eau est un solvant "dispersant".
Dans le vide: Fvide
=![]()
Dans l'eau: Feau=![]() La
force coulombienne y est divisée par 80, toutes choses égales
par ailleurs, par rapport à la force coulombienne dans le vide:
les ions y sont mieux "dispersés" par conséquent.
La
force coulombienne y est divisée par 80, toutes choses égales
par ailleurs, par rapport à la force coulombienne dans le vide:
les ions y sont mieux "dispersés" par conséquent.
Avec e=e0.er
et
e0,
permittivité du vide, @ 8.85.10-12
F.m-1.
Retour
au plan du cours sur les pH des solutions.
I.2. Propriétés physico-chimiques de l'eau: les ions de l'eau pure. L'autoprotolyse de l'eau.
Retour au plan du cours sur les pH des solutions aqueuses.
Du fait de l'agitation thermique il y a dans le solvant H2O des collisions intermoléculaires qui, pour certaines d'entre elles, sont efficaces et conduisent à des ruptures de liaisons de covalence :
A 25°C on a (H3O+) = (HO-) = 10-7 mol.L-1.
REM 1: dans l'eau il y a
Pour mesurer le degré d'ionisation de l'eau on utilise un "conductimètre" (voir en T.P.)
L'eau est un conducteur, même faible, du courant électrique, et a pour résistance R.
Il existe la relation R =
On pose r =1/g, avec g conductivité de l'eau. La conductivité g est exprimée alors en siemens par mètre, symbole S.m-1.
On trouve que la conductivité de l'eau pure à 25°C est égale à 5.5.10-6 S.m-1 et que sa résistivité vaut: 1.81.105 W.m .
C'est la mesure de la conductivité de l'eau pure, fonction de la concentration des ions hydronium et des ions hydroxyde présents, qui permet de trouver la valeur de 10-7 mol.L-1 pour chacune de ces concentrations.Voir TP de conductimétrie, en fin de site WEB. Le produit de leurs activités donne la valeur 10-14.
On caractérise alors l'équilibre 2H2O
REM 2: il n'y a pas d'unités pour Ke.
On multiplie ce qu'on appelle des "activités" et non des concentrations, exprimées par exemple en mol.L-1. Les concentrations "classiques" sont à cet effet multipliées par un coefficient sans unité, appelé "coefficient d'activité", et divisées par la concentration "standard", de référence, prise égale à CO = 1 mol.L-1.
Alors on "comprend" l'absence d'unités pour les activités: voir suite du cours en "thermodynamique chimique".
REM3: Ke varie avec la température selon la relation:
A 10°C (283K) la valeur de pKe est de 14.53logKe = - -3.55, avec T en kelvins.
A 20°C (293K) la valeur de pKeest de 14.16
A 30°C (303K) la valeur de pKeest de 13.83
A 60°C (333K) la valeur de pKeest de 13.02
On voit que si T augmente alors les chocs thermiques deviennent plus efficaces et Ke augmente, d'où pKe diminue.
Retour au plan du cours sur les pH des solutions aqueuses.
Retour au plan du cours sur les pH des solutions aqueuses.
On a la relation: pH =
-log(H3O+)
= colog(H3O+) = -![]() ,
ou, sous forme exponentielle: (H3O+) = 10-pH.
,
ou, sous forme exponentielle: (H3O+) = 10-pH.
Cette définition a été
donnée par le Danois
SØRENSEN en 1909.
REM: Le pH égal à 7de l'eau pure à 25°C, une douce fiction....
Le pH de l'eau pure est plus souvent proche de 6, voire de 5, en raison de l'extrême difficulté à obtenir de l'eau "pure" justement. Le dioxyde de carbone CO2 dissous dans l'eau acidifie cette dernière, d'où le pH différent de 7....
Que faire pour purifier de l'eau?
1. Distillation classique:
voir la célèbre malette de jeux "Chimie 2000". Procédé
coûteux
en
énergie.
2. Utilisation de résines
échangeuses d'ions, cations puis anions, afin
d'obtenir de l'eau "déminéralisée".
Insérer schéma
des résines anioniques et cationiques.
Les résines échangeuses
d'ions, comment ça marche?
On fait passer de l'eau salée,
mélange de molécules d'eau, d'ions Na+ et d'ions
Cl-, sur la résine I, échangeuse de
cations.
Il y a échange des ions
Na+ avec les ions H3O+.
On reprend le liquide obtenu à
la sortie de la résine I et on le fait passer sur la résine
II, échangeuse d'anions.
Il y a échange entre
les ions Cl- et les ions HO-.
Bilan: on récupère à la sortie des résines un mélange de molécules d'eau, d'ions hydronium (oxonium) et d'ions hydroxyde.
Or, comme ces deux catégories d'ions sont en quantités identiques on a la réaction bien connue suivante:
L'intérêt de ces résines est d'obtenir de l'eau desionisée,déminéralisée, un peu comme une eau "distillée" mais sans avoir besoin de chauffer. Cela permet de faire des économies d'énergie. C'est cette eau qui est vendue en grandes surfaces à usage des fers à repasser, etc....
3. Osmose inverse.
Le procédé par osmose
inverse est très utilisé actuellement dans le dessalement
à grande échelle de l'eau de mer dans des pays tels que l'Arabie
saoudite.
L'osmose "directe" est un
phénomène que vous avez dû étudier au lycée
en cours de Sciences naturelles.
Lorsqu'on met au contact l'une
de l'autre, par l'intermédiaire d'une paroi semi perméable
(laissant passer les molécules de solvant mais pas de soluté),
une solution et un solvant pur, il s'établit au bout d'un certain
temps, une "surpression" au sein de la solution qui s'oppose à ce
que les molécules de solvant pur pénètrent dans la
solution.C'est ce qu'on appelle la pression osmotique de la solution.
Si l'on exerce sur une solution
une pression supérieure à la pression osmotique on peut alors
réaliser ce qu'on appelle l'osmose inverse, c'est à dire
que le solvant de la solution passe dans le compartiment du solvant pur.
C'est ainsi qu'on obtient une eau désalinisée.
Retour plan du cours sur les pH des solutions aqueuses.
Retour au plan du cours sur les pH des solutions aqueuses.
II.1. Quelques définitions au cours de l'Histoire....
Retour au plan du cours sur les pH des solutions aqueuses.
II.1.a.Théorie de Svante ARRHENIUS,Suédois, Prix NOBEL de Chimie en 1903, le troisième Prix NOBEL à être décerné par l'Académie suédoise.
Qu'est ce qu'un "acide"?
C'est une espèce chimique susceptible de libérer des ions
H+.
Qu'est ce qu'une "base"?
C'est une espèce chimique capable de libérer des ions HO-,
écrit OH-à l'époque et ce, jusqu'à
très récemment....
Cette théorie qui venait à la suite de la théorie des ions, justement élaborée par ARRHENIUS, ce pour quoi il avait reçu son Prix NOBEL, ne faisait pas la place au rôle pourtant majeur (mais ça on ne le saura qu'après) du solvant. Elle ne permettait pas, par exemple, de considérer que l'ammoniaque, alias solution aqueuse du gaz ammoniac, de formule NH3, était une "base", ce qu'elle était par ailleurs.
II.1.b.Théorie de BRONSTED et LOWRY (1923).
Qu'est- ce qu'un "acide"? C'est une espèce chimique capable de libérer des ions H+.
Qu'est-ce qu'une "base"? C'est une espèce chimique capable de capter des ions H+.
Exemple:
HA est "l'acide conjugué" de la base A-.
On dit que HA et A- forment un "couple acido-basique". On le notera HA/A-
On donnera plusieurs exemples, sans
trop insister, un ou deux et puis, on a compris...
Il est convenu de mettre en premier
l'espèce acide et en second la base conjuguée de cet acide.
Couple acide éthanoïque /ion éthanoate:CH3CO2H/CH3CO2-
Couple acide phosphorique
/ion
dihydrogénophosphate:
H3PO4/H2PO4-
Couple dioxyde de carbone en
solution aqueuse /ion hydrogénocarbonate:
H2CO3
ou CO2 (aq )/ HCO3-
Couple ion ammonium /ammoniac:
NH4+/NH3
.
II.1.c. Théorie de LEWIS (1923).
Cette théorie sera surtout utilisée en DEUG 2 ème année, en Chimie organique
Qu'est ce qu'un "acide"?
C'est une espèce chimique qui possède une lacune électronique sur sa couche externe.
C'est l'exemple du chlorure d'aluminium
AlCl3qui ne satisfait pas à la "sacro sainte"
règle
de l'octet.
Il n'y a que 6 électrons
sur la couche externe au lieu des 8 si j'ose dire "attendus".
C'est l'exemple du trifluorure de bore BF3, qui lui aussi n'a que 6 électrons sur sa couche externe, au lieu des huit "attendus".
C'est enfin l'exemple de l'ion hydrogène, le proton H+, qui n'a pas sa couche externe saturée à deux électrons, et pour cause, puisqu'il n'a pas.... d'électrons tout court!
Qu'est ce qu'une "base"?
C'est une espèce chimique qui est porteuse sur sa couche externe d'un doublet électronique susceptible d'interagir avec une lacune électronique.
C'est le cas de la molécule d'eau H2O, c'est aussi celui de la molécule NH3.
Qu'est-ce qu'une réaction acidobasique au sens de LEWIS?
C'est l'exemple de la réaction
entre le chlorure d'aluminiumAlCl3 et l'ammoniacNH3.
Il se forme ce que l'on appelle
un "adduit" de
LEWIS, de formule Cl3Al-N+H3
Il existe alors une charge négative sur l'atome d'aluminium : pour qu'il y ait liaison covalente entre l' atome d' aluminium et l' atome d'azote, l'azote donne un électron de son doublet à l'atome d'aluminium , neutre au départ. En gagnant un électron l'atome d'aluminium se charge négativement d'où l'apparition de la charge indiquée.
Il existe, par conséquent, une charge positive sur l'atome d'azote. En effet, en donnant un électron de son doublet à l'atome d'aluminium afin de créer une liaison de covalence, l'atome d'azote se charge positivement, d'où l'apparition de la charge indiquée.
Bien entendu l'adduit de LEWIS est ici neutre électriquement.
Ces notions seront largement exploitées en chimie organique où l'on décrit convenablement de très nombreux mécanismes réactionnels comme étant composés de nombreuses réactions de LEWIS: je pense notamment au cas des réactions de substitution électrophile du type FRIEDEL CRAFTS.
Retour au plan du cours sur les pH des solutions aqueuses.
II.2.Les acides et les bases dans l'eau: développement des applications de la théorie de BRONSTED.
Retour au plan du cours sur les pH des solutions aqueuses.
II.2.a. Les couples acidobasiques de l'eau.
II.2.a.a.L'eau
H2O est une "base" de BRONSTED.
Soit la réaction entre le
chlorure d'hydrogène
HCl et l'eau H2O.
Base 1 Acide 2 Acide 1 Base 2
II.2.a.b. L'eau H2O est un "acide" de BRONSTED.
Soit la réaction entre l'ammoniac NH3 et l'eau H2O.
| H2O + | NH3 | NH4+ + | HO- | |
|
|
|
|
|
Deux couples acidobasiques sont mis en évidence : le couple H2O/HO-et le couple NH4+/NH3.
Conclusion: les deux couples acidobasques de l'eau solvant sont donc:
H3O+/H2O
et H2O/HO-.
II.2.a.g.
L'eau H2O est amphotère.
En raison de ce qui précède
on dit alors que l'eau H2O est un "amphotère"
ou un "ampholyte" , à la fois "acide" donc ,
et "basique".
Comparons ces deux couples au moyen de leur constante d'acidité Ka.
Couple H3O+/H2O:
On a la réaction d'échange
de proton H+ d'une molécule d'eau à l'autre:
On écrit la constante d'acidité Ka associée à cet équilibre de la façon suivante:
Keq =
 Puisque
les deux concentrations sont égales, vu qu'elles décrivent
la concentration des ions H3O+ d'un même
milieu,
on a Keq= 1. D'où, bien évidemment pKeq=
0.
Puisque
les deux concentrations sont égales, vu qu'elles décrivent
la concentration des ions H3O+ d'un même
milieu,
on a Keq= 1. D'où, bien évidemment pKeq=
0.
On en déduit que le pKa du couple H3O+/H2O est alors égal à 0.
Couple H2O/HO-:
On a la réaction d'autoprotolyse de l'eau qui intervient:
H2O + H2O
Acide 1 Base 2 Acide 2 Base 1
On peut alors écrire Keq=
On met 1 au dénominateur car l'activité du solvant H2O est considéré comme étant égale à 1.
Cela veut dire qu'on est en solution "infiniment diluée" si j'ose dire, et que l'eau est "pure", c'est à dire qu'il n'y a que des molécules H2O dedans.
Ce qui est bien proche de la vérité.
La constante de cet équilibre , Keqest égale à Ka, c'est à dire ici à Ke, la constante d'autoprotolyse de l'eau , pour une température donnée.
On aura donc , à 25 °C, Ke = 10-14, d'où, bien sûr, pKe =14.
Le pKa du couple H2O/HO-est donc égal à 14.
Conclusion : l'acide le plus fort dans l'eau sera H3O+ et la base la plus forte sera HO-.
A partir de là on peut dire , de façon équivalente, que si l'acide le plus fort dans l'eau est H3O+, alors l'acide le plus faible dans l'eau sera... L'eau.
De même, la base la plus forte dans l'eau sera HO-, mais la base la plus faible dans l'eau sera... l'eau.
On dit également que l'eau a un effet "nivelant".
On voit écrit dans certains ouvrages que le pKa de l'acide chlorhydrique, par exemple, est négatif.
Est-ce une "erreur"?
Non, cela veut dire que dans
certains solvants, tel l'acide éthanoïque concentré
et pur, le "glacial acetic acid" des Anglo-Saxons, on a l'acide
chlorhydrique HCl qui est un acide partiellement ionisé.C'est
aussi le cas de l'acide bromhydrique, etc...On peut différencier
leurs forces respectives à l'aide d'un pKa dans ces solvants
autres que l'eau. Voir l'ouvrage de M. DEFRANCESCHI cité en référence.
Or, dans l'eau ,
HCl est
totalement ionisé et on ne le différencie
nullement d'autres acides tels que l'acide bromhydrique HBr
ou l'acide
iodhydrique HI.
L'eau a joué son rôle
de solvant "nivelant" quant à l'acidité de tous ces
hydracides.
Ils sont tous "égaux" dans l'eau si j'ose dire...
Il existe par exemple dans d'autres
solvants que l'eau des bases telles que l'anion éthanolate,
de formule CH3CH2O- . On le fabrique
à partir d'éthanol anhydre, CH3CH2OH
et de sodium, Na. Le solvant de la réaction est l'éthanol
anhydre lui même. Il se forme de l'éthanolate de
sodium (à ne pas confondre avec l'éthanoate de sodium,
de formule NaCH3CO2 !) et il se produit un
dégagement
de
dihydrogène
gazeux H2.
Si l'on met au contact de l'eau
l'anion
éthanolate
il se produit alors la réaction
:
On ne peut donc pas trouver d'anion éthanolate dans l'eau.
La constante d'équilibre
de la réaction précédente est, en effet, très
en faveur des produits.
L'eau a joué son rôle
nivelant dans ce phénomène.
On peut également en déduire que dans le solvant eau tous les acides qu'on trouvera auront forcément un pKa compris entre 0 et 14.
Si l'on trouve dans la littérature que le pKa des alcools est de l'ordre de 18, cela veut dire que dans l'eau il ne pourra jamais y avoir comme réaction la réaction suivante:
CH3CH2OH
+ H2O ![]() H3O+ + CH3CH2O-
H3O+ + CH3CH2O-
Revoyez ce que j'avais souligné sur la théorie de Svante ARRHENIUS et vous verrez qu'elle ne parlait jamais de ce rôle, somme toute majeur, du solvant.
On n'accablera pas ici ARRHENIUS car, c'est là le rôle d'une science, c'est de toujours évoluer.
II.2.b. Les autres couples acidobasiques
dans l'eau solvant.
On prendra le cas d'un monoacide
faible, c'est à dire le cas "général", HA.
On a la réaction suivante:
Avec Ka
= ![]()
L'activité n'a pas d'unités. On a la relation suivante:
(H3O+) =
On a gqui est un nombre positif, sans dimension, appelé "coefficient d'activité", qui tend vers la valeur 1 quand la concentration de H3O+ tend vers 0.C'est le cas idéal des solutions infiniment diluées.
Le coefficient d'activité g se calcule (voir suite du cours de Thermodynamique chimique) en tenant compte de la concentration des ions présents dans la solution, ainsi que du carré de leur charge électrique : c'est ce qu'on appelle le calcul de la force ionique I d'une solution qui peut conduire à l'application de la méthode, dite de DEBYE-HUCKEL, pour ladite détermination du facteur d'activitég.
C0 désigne la concentration standard , de référence, prise égale à 1 mol.L-1.
Pour simplifier, on n'indiquera plus qu'on divise toutes les concentrations par C0 = 1 mol.L-1, et on estimera que le coefficient gest proche de la valeur 1.
Alors, et à ce titre seulement, activité et concentration seront mesurées par les mêmes chiffres, mais l'activité est sans dimension, alors que la concentration oui.
A partir de Ka, on peut prendre le logarithme décimal de son expression. On arrive très facilement à:
pH = pKa
+ log ![]()
On peut également définir une constante dite de "basicité" du couple HA / A-.
Kb
= 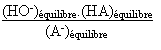
Ka.Kb = (H3O+).(HO-) = Ke
C' est là une relation importante, qu'on note souvent sous sa forme logarithmique:
pKa + pKb = pKe = 14, à 25 °C
Remarque importante: les tables de constantes physico-chimiques ne donnent, en général, jamaispKb. On comprend aisément qu'on peut retrouver pKb , connaissant pKa, moyennant une opération tout à fait élémentaire.
Comparons les forces respectives de deux acides faibles, afin de voir l'influence du pKa sur leur état physique en solution aqueuse, toutes choses étant égales par ailleurs.
Prenons chacun de ces acides, séparément,
à la concentration C0 de 0.1
mol.L-1.
Les pKa respectifs
des acides "méthanoïque" (formique) et "éthanoïque"
(acétique) sont, à 25 °C, 3.80 et 4.75.
Nous avons la réaction générale suivante:
HA + H2O ![]() H3O+
+ A-
H3O+
+ A-
Dans un litre de solution,
nous avons:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
J'ouvre ici une grosse parenthèse, qui s'impose vu ce que je viens d'écrire:
En effet, on considèrera
qu'on peut négliger
les ions H3O+apportés
par l'eau.
On appliquera ce qu'on appelle
la "réaction prépondérante".
Cette méthode, bien
que n'étant
absolument pas la seule, permet d'approcher la
question des équilibres chimiques avec une certaine aisance.
Elle demande essentiellement deux choses au départ:
Il faut rechercher dans le milieu
étudié quelle est la
valeur la plus élevée
des
constantes
d'équilibre
censées décrire les réactions
possibles intervenant réellement dans le milieu considéré.
Ici, on voit par exemple que la
constante d'acidité Ka de chacun des acides, formique et acétique,
est notoirement plus élevée que la valeur de la constante
d'équilibre d'autoprolyse de l'eau.
On a donc 10-3.8
et 10-4.75 qui sont beaucoup plus grandes que 10-14.
Il faut ensuite voir par le calcul
si la quantité de matière produite par chacune de ces
réactions censées être "prépondérantes"
est quantitative ou pas.
On verra ci après
que la mise en solution de l'acide formique
à
0.1 mol.L-1
donnera naissance à 3.890.10-3mol d'ions H3O+par
litre de solution préparée.
Cette quantité de matière
est
très
grandedevantles quelque 10-7 mol d'ions H3O+
fournis
par l'autoprotolyse de l'eau.
On a donc, dès lors,
grandement
le droit de négliger les ions H3O+provenant
de l'eau, et c'est pourquoi on a mis dans le tableau
précédent
la valeur "0" au temps initial
pour la valeur, exprimée
en mole, des ions H3O+.
Cette remarque est la même
pour l'acide acétique
(voir calculs ci après).
Remarque :
si jamais on avait une réaction prépondérante dont
le bilan de matière apparaissait comme négligeable devant
celui apporté par d'autres réactions chimiques, alors il
faudrait changer de réaction prépondérante.
On verra cela dans les
exercices du cours.
Revenons à notre exercice après cette parenthèse de taille : la constante Kas'écrit donc, dans chaque cas :
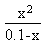
Pour l'acide méthanoïque ( formique) on trouve que x = 3.890.10-3 mol.L-1.
Pour l'acide éthanoïque (acétique) on trouve que x = 1.325.10-3 mol.L-1.
On posera a"degré de dissociation" d'un acide, le rapport a(exprimé en pourcentage) suivant:
On dira alors: toutes choses
étant égales par ailleurs, plus le pKa
d'un acide sera faible
plus l'acide sera dissocié,
donc
fort.
De même, a contrario,
plus le pKa d'un acide sera élevé,
alors moins l'acide sera
dissocié, plus l'acide
sera faible.
Ici, avec des degrés de dissociation a de respectivement 3.89% et 1.33% on peut dire que l'acide méthanoïque est plus fort que l'acide éthanoïque. Encore une fois, toutes choses égales par ailleurs.
En effet, si l'on prend ces deux acides, mais à deux concentrations différentes, par exemple 0.1 mol.L-1 pour l'un et 0.2 mol.L-1 pour le second, on ne peut rien conclure à la seule vue du degré de dissociation a. On ne peut comparer que des choses comparables.