II.3.e.
Cas des polyacides. Les pH de leurs solutions aqueuses.
Retour
au plan du cours sur les pH des solutions aqueuses.
Les polyacides sont très répandus dans la
nature: on citera le cas de l'acide citrique, un triacide organique fondamental,
ne serait-ce que par son intervention dans le métabolisme des glucides
en CO2 et H2O selon le cycle de KREBS, connu
également sous le nom de cycle de l'acide "tricarboxylique" ou de
"l'acide citrique" ; métabolisme qui intéresse chacun d'entre
nous.
On citera encore le cas de l'acide phosphorique, H3PO4,
qui entre dans les mécanismes biochimiques de transport et d'échange
d'énergie: passage de l'ATP (non ce n'est pas Roland GARROS qui
vous parle!!! [rires!!!] ) à l'ADP, avec libération
d'énergie et création d'un groupement phosphate.Dans l'ADP
et l'ADP, respectivement "adénosine triphosphate" et "adénosine
diphosphate" , l' acide phosphorique est présent sous la forme d'anhydride
minéral: la rupture des liaisons P-O-P est fortement libératrice
d'énergie.
On citera comme polyacide très connu, le cas du
dioxyde de carbone CO2, dont la solution aqueuse est appelée
"acide carbonique", de formule H2CO3 (formule
par ailleurs "contestée"), ou CO2 aq.
On pourra citer aussi le dioxyde de soufre SO2,
dont l'oxydation par le dioxygène de l'air, en présence d'humidité
(eau...) est responsable des pluies acides, composées d'un autre
diacide, l'acide sulfurique, de formule H2SO4. La
solution aqueuse de dioxyde de soufre s'appelle l' acide sulfureux. On
la note, soit "H2SO3", soit "SO2aq".
On citera pour finir (?) le cas d'un composé qui
ne sent pas très bon et qui fait le bonheur des farceurs, je veux
parler du sulfure de dihydrogène, de formule H2S, plus
connu sous le nom d'hydrogène sulfuré (vieux nom), qui sent....
les oeufs pourris, et que Nino FERRER a si bien décrit dans une
de ses plus belles chansons à mon avis, "La maison près de
la fontaine" (1972, mes 12 ans!!!!!). La solution aqueuse de sulfure de
dihydrogène s'appelle l'acide sulfhydrique.
On comprend DONC, le très grand intérêt
que revêt l'étude des solutions aqueuses de polyacides.
On commencera tout d'abord par un exemple, celui d'un
diacide d'origine organique, le plus simple des diacides organiques, l'acide
oxalique, de formule HO2C-CO2H, abrégé
en H2C2O4 ou, ici, par commodité
de dactylographie, par "H2A".
L'acide oxalique possède deux pKai
: 1.2 et 4.3.
Les équilibres chimiques relatifs à ces
deux constantes d'acidité sont les suivants:
HO2C-CO2H + H2O 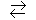 HO2C-CO2-
+ H3O+
L' équilibre est caractérisé par une
constante d'acidité Ka1 de valeur: 0.063, c'est
à dire 10-1.2.
HO2C-CO2-
+ H3O+
L' équilibre est caractérisé par une
constante d'acidité Ka1 de valeur: 0.063, c'est
à dire 10-1.2.
Son expression littérale sera alors: Ka1
= 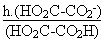 HO2C-CO2-
+H2O H3O+
+ -O2C-CO2-
L'équilibre est caractérisé par une
constante d'acidité Ka2 de valeur 5.10-5,
c'est à dire 10-4.3.
HO2C-CO2-
+H2O H3O+
+ -O2C-CO2-
L'équilibre est caractérisé par une
constante d'acidité Ka2 de valeur 5.10-5,
c'est à dire 10-4.3.
Son expression littérale sera alors: Ka2
= 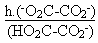
On notera l'anion hydrogéno-oxalate HO2C-CO2-
par "HA-" et l'anion oxalate -O2C-CO2-
par "A2-".
On aura certainement besoin de connaître et de
tracer le diagramme de prédominance des espèces dissoutes
de l'acide oxalique en solution aqueuse.
On écrira tout d'abord l' équation de conservation
de la masse:
C0 = (H2A)équilibre
+ (HA-)équilibre + (A2-)équilibre
.
De la constante Ka1 on tire: (H2A)
= 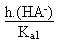
De la constante Ka2 on tire (A2-)
= 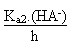
Enfin, on écrit la trivialité suivante:
(HA-) = (HA-)
On pose: h =10-pH , Ka1 =
10-pKa1 et Ka2 = 10-pKa2.
En faisant la somme des termes de gauche d'un côté,
la somme des termes de droite d'un autre côté, on arrive à
ne plus exprimer (HA-) qu'en fonction de C0, du pH,
de pKa1 et de pKa2.C'est bien ce qu'on cherchait...
On arrive dès lors à:
(HA-) = 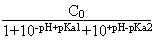 Puis, en multipliant ce résultat par 10-pH+pKa1,
on arrive à:
(H2A) =
Puis, en multipliant ce résultat par 10-pH+pKa1,
on arrive à:
(H2A) = 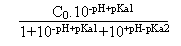 Enfin, en multipliant l'expression de (HA-) par
10+pH-pKa2, on arrive à:
(A2-) =
Enfin, en multipliant l'expression de (HA-) par
10+pH-pKa2, on arrive à:
(A2-) = 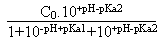 A partir de là on peut tracer, en fonction du pH,
de pKa1 et de pKa2, le diagramme de prédominance
des espèces dissoutes de l'acide oxalique.
A partir de là on peut tracer, en fonction du pH,
de pKa1 et de pKa2, le diagramme de prédominance
des espèces dissoutes de l'acide oxalique.
Par commodité on tracera plutôt, pour chacune
de ces formes, le pourcentage de chacune en fonction du pH, de pKa1
et de pKa2. Il suffit par rapport aux formules précédentes
de diviser par C0 et de multiplier le résultat par 100,
pour l'avoir en pourcentage.
Ces formules sont ici établies pour l'acide oxalique
mais elles resteront vraies pour tout diacide faible H2A . Il
suffira de changer les valeurs des pKai .
Diagramme de prédominance
de l'acide oxalique.

On remarque, bien évidemment, que si le pH augmente
la forme H2A diminue de façon monotone, et que la forme
A2- augmente, quant à elle, de façon également
monotone.
Quant à la forme HA- elle augmente,
passe par un maximum puis diminue.
Le maximum de la forme HA- est atteint (voir
calcul de dérivée en maths) pour la valeur de pH égale
à 0,5.(pKa1 + pKa2).
Comment calculer le pH des diacides?
Il y a deux méthodes.Deux méthodes au moins....
La première nécessite la résolution
de l'équation d'électroneutralité.
Cette équation présente au niveau de son
écriture une très grosse difficulté qui tient, en
fait de l'énorme paradoxe plutôt.
L'équation complète d'électroneutralité
pour tout diacide H2A est en effet égale à:
h = w + (HA-)
+ 2.(A2-)
Je vais vous donner la démonstration du "2" devant
(A2-) à l'aide de mon animal favori, ou plutôt
de l'un de mes animaux favoris, le "nounours", le "bear", le "bär",
el "oso", si vous préférez.
J'utiliserai, pour la commodité de l'exposé,
des nounours: à deux oreilles (ce sera H2A), à
une oreille (ce sera HA-) et, enfin sans oreille, ce sera alors
A2-. Les ions hydronium seront symbolisés par une petite
oreille de nounours.

|


 |
| Avant instauration des équilibres acidobasiques |
Après instauration de tous les équilibres
acidobasiques |
Si l'on écrit l'équation d'électroneutralité,
à savoir:
h = (HA-) + a.(A2-)
et que l'on remplace h, (HA-) et (A2-)
par leurs valeurs respectives, à savoir 5, 3 et 1, on voit que
5 = 3 + 2.1
d'où a vaut 2.Avec
un 2 devant (A2-) on obtient le bon bilan électrique.
L'équation d'électroneuralité est
donc définitivement décrite par:
h = (HA-) + 2.(A2-)
Bien sûr, dans l'équation d'électroneutralité
on a négligé tout de suite w
devant h car, bien entendu, le pH d'un diacide est... acide.
L'histoire du "2" peut bien sûr se généraliser
au degré "n".
Chaque fois que dans une équation d'électroneutralité
on a une activité d'un ion dont la charge est différente
de l'unité on devra mettre devant l'activité un coefficient
a
qui sera égal à la valeur absolue de la charge de l'ion.
Pour ce qui est du calcul du pH maintenant, on se
placera d'emblée dans le seul cas qui soit intéressant, à
savoir le cas où la concentration initiale C0 du diacide
H2A sera supérieure à 10-6 mol.L-1.Une
telle concentration limite, pour un acide de masse molaire moyenne disons
de 100 g.mol-1, est de 0.1 mg.L-1. Si vous
pesez une telle quantité sur une balance de précision vous
verrez que 0.1 mg ce n'est rien d'autre que l'équivalent d' une
poussière qui sera dissoute dans 1 litre d'eau. C'est ce que l'on
appelle une teneur de "100 ppb", c'est à dire de 100 microgrammes
par kilogramme de matière. On est donc bien à 10-6
mol.L-1 à la limite du raisonnable dans le calcul des
pH. Ce que je n'ai pas arrêté de dire depuis le début
de ce cours "on line".
L'équation d'électroneutralité se
simplifie donc, vu ce que j'ai fait comme approximation plus que raisonnable,
en:
h = (HA-) + 2.(A2-)
Si l'on remplace (HA-) et (A2-)
par les expressions en fonction de h, de Ka1 et de Ka2
précédentes, on arrive à un polynôme en h de
degré supérieur certainement à 2.
Aujourd'hui, à l'heure des calculateurs puissants,
résoudre par itération (à moins que par hasard vous
ne "deviniez" la valeur qu'il faut trouver) une telle équation est
un jeu d'enfant. Il suffit juste de bien paramétrer son logiciel
de calcul et on arrive par dichotomie à trouver assez rapidement
la valeur de h, partant la valeur de pH, qui vous "inquiète"
ou qui vous "amuse"....
L'autre méthode fait appel à l'utilisation
de la (ou des) réaction(s) prépondérante(s).
Examinons le cas du calcul du pH d'une solution aqueuse
d'acide oxalique à 0.05 mol.L-1 par exemple.
Si de diacide était fort, c'est à dire
totalement dissocié, on aurait pour cette solution un pH de 1. En
effet, 0.05 mole d'acide oxalique libèrerait le double en ions hydronium.
La concentration de ces ions serait égale alors à 0.1 mol.L-1.
Le pH de la solution ici sera donc compris forcément
entre les bornes 1 et 7.
Ecrivons de façon abrégée le premier
équilibre chimique qui a lieu :
H2A + H2O H3O+
+ HA-
On a écrit le premier équilibre car c'est lui
qui est, a priori, la réaction prépondérante.
En effet, c'est Ka1 qui est ici la plus
grande des constantes d'équilibre. Première partie du raisonnement.
Ensuite c'est bien dans le premier équilibre qu'intervient
la seule espèce susceptible de former des ions H3O+
en quantité notable, à savoir H2A.Deuxième
partie du raisonnement.
A l'instant initial, vu qu'on négligera les ions
de l'eau, on écrira, pour un litre de solution, le nombre de moles
de chacun des composés présents, l'eau excepté puisque
c'est le solvant de la réaction.
A l'équilibre chimique, au bout d'un temps infini
on notera les quantités de chacune des espèces en présence.
Les résultats, exprimés pour un litre de
solution, sont donnés dans le tableau suivant:
|
H2A |
H3O+ |
HA- |
| temps initial |
0.05 mol |
0 mol |
0 mol |
| temps infini |
0.05-x mol |
x mol |
x mol |
On remplace dans l'expression littérale de Ka1
chacune des concentrations, ramenées à des activités,
par leur expression figurant dans le tableau ci-dessus.
On obtient l'équation suivante:
Ka1 =  Si H2A n'avait été qu'un monoacide
faible, il suffisait de résoudre l'équation du second degré
en x obtenue ici et c'en était fini du pH de l'acide faible.
Si H2A n'avait été qu'un monoacide
faible, il suffisait de résoudre l'équation du second degré
en x obtenue ici et c'en était fini du pH de l'acide faible.
Or, ici, il faut tout d'abord calculer la valeur de x
puis ensuite vérifier si le second équilibre n'est pas susceptible,
lui aussi, de fournir des ions H3O+ de façon
quantitative, afin de modifier le pH.
Tous calculs faits, vu que Ka1 vaut environ
0.063, on est amené à résoudre l'équation du
second degré suivante:
x2 + 0.063.x -0.0032 = 0
On trouve, comme seule valeur physiquement acceptable, la
valeur x = 0.033
Ce veut dire, si un seul équilibre, caractérisé
par Ka1, existait, on aurait comme composition du milieu à
t infini:
(H3O+) = 0.033 mol.L-1
(HA-) = 0.033 mol.L-1
(H2A) = 0.017 mol.L-1
Or, le second équilibre entre en jeu maintenant.
Une partie de HA- est en effet susceptible
de créer, à son tour cette fois, un peu d'ions hydronium,
ce qui modifierait la valeur finale du pH de la solution.
Voici ce qui se passe entre le moment où le premier
équilibre se terminerait, s'il était seul, et la composition
finale réelle de la solution.
On présentera les résultats, toujours donnés
en mol.L-1, dans un tableau:
|
HA- |
H3O+ |
A2- |
| temps t, à la fin de l'instauration du premier équilibre,
s'il était seul à intervenir |
0.033 mol |
0.033mol |
0 mol |
| temps final, lorsque tous les équilibres se sont instaurés |
0.033-x mol |
0.033 +x mol |
x mol |
On aura à résoudre l'équation du second
degré obtenue à partir de:
Ka2 = 5.10-5 =  L'équation du second degré s'écrit
alors:
x2 + 0.033.x -1.67.10-7 = 0
On résout et la seule solution physiquement acceptable
conduit à x = 1.096.10-3
L'équation du second degré s'écrit
alors:
x2 + 0.033.x -1.67.10-7 = 0
On résout et la seule solution physiquement acceptable
conduit à x = 1.096.10-3
Ainsi donc, le second équilibre chimique a conduit
à la fabrication de 1.096.10-3 mol d'ions H30+
en plus des 0.033 mol créées par le premier.
La quantité totale d'ions hydronium présente
par litre de solution aqueuse est donc égale à la somme de
ces deux quantités. On trouve: 0.034 mol.L-1.
On voit que c'est la première réaction,
caractérisée par la constante d'équilibre Ka1
qui crée la majorité des ions hydronium de la solution et
donc impose le pH. Le correctif est minime. On peut pratiquement considérer,
mais après coup seulement, que l'acide oxalique se comporte, à
cette concentration, comme un monoacide faible de pKa égal
à pKa1.
Cela n'était pas évident a priori. Il
fallait le montrer.
Le pH de la solution d'acide oxalique à 0.05 mol.L-1
est donc égal à 1.47.
Cette méthode peut se généraliser
à tout diacide faible mis en solution dans l'eau.
Pour l'acide tartrique, dont les deux pKa sont respectivement:
3.13 et 4.31 , pour une concentration initiale C0 égale
à 0.1 mol.L-1 on trouve un pH de 2.08 et une contribution négligeable
de la seconde acidité (4.8.10-5 mol d'ions H3O+
apportés
par la seconde acidité, contre 8.23.10-3 par la première).
A 0.01 mol.L-1 la contribution de la seconde
acidité à la valeur du pH de la solution vaut environ 4%
(4.78.10-5 mol d'ions hydronium contre 0.0024 apportés
par la première acidité). On trouve un pH de 2.61.
Les autres exemples seront donnés lors des
exercices d'application.
Prenons l'exemple des triacides désormais.
On prendra les cas de l'acide phosphorique, de formule
H3PO4 et celui de l'acide citrique.
On notera le triacide de façon générique
"H3A".
On aura, à chaque fois, les constantes d'acidité
Kai suivantes:
Ka1 = 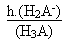
Ka2 = 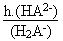
Ka3 = 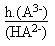 A partir de l'équation de conservation de la matière,
C0 = ( H3A) + (H2A-)
+ (HA2-) + (A3-)
en reprenant le raisonnement suivi pour les diacides on arrive
à établir les pourcentages des quatre espèces dissoutes
provenant du triacide:
(H3A) =
A partir de l'équation de conservation de la matière,
C0 = ( H3A) + (H2A-)
+ (HA2-) + (A3-)
en reprenant le raisonnement suivi pour les diacides on arrive
à établir les pourcentages des quatre espèces dissoutes
provenant du triacide:
(H3A) = 
(H2A-) = 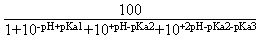
(HA2-) =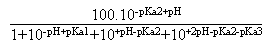
(A3-) =  A partir de là on peut tracer le diagramme des espèces
dissoutes de chacun des triacides.
A partir de là on peut tracer le diagramme des espèces
dissoutes de chacun des triacides.
Pour l'acide phosphorique, de pKai respectifs
2.15, 7.2 et 12.1 on aura:
Diagramme de prédominance
de l'acide phosphorique.

Ce diagramme pourra nous servir si l'on veut résoudre
l'équation d'électroneutralité afin d'accéder
au pH de la solution.
Pour l'acide citrique, de pKai respectifs 3.1,
4.8 et 6.4 on aura:
Diagramme de prédominance
de l'acide citrique.

Revenons à l'acide phosphorique
et calculons le pH d'une solution de concentration 0.05 mol.L-1.
Nous emploierons tout d'abord la
méthode de la réaction prépondérante.
En reprenant la méthode de
la réaction prépondérante on voit qu'on a comme réaction
prépondérante la réaction caractérisée
par la constante d'équilibre Ka1.
On a dès lors à résoudre
l'équation du second degré suivante, sachant que pKa1
vaut 2.15 et que C0 vaut 0.05 mol.L-1
x2 + 7.08.10-3.x -3.54.10-4
= 0
D'où x = 0.0156 .
Si l'on recherche la réaction prépondérante
secondaire on voit qu'elle correspond à la réaction de constante
d'équilibre égale à Ka2.
On est amené alors à résoudre :
10-7.2 = 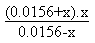 Soit, tous réarrangements opérés,
l'équation du second degré suivante:
x2 + 0.0156 -9.8.10-10
= 0
On résout et on trouve x = 6.2.10-8.
Soit, tous réarrangements opérés,
l'équation du second degré suivante:
x2 + 0.0156 -9.8.10-10
= 0
On résout et on trouve x = 6.2.10-8.
On voit tout de suite que la seconde réaction
ne produit pas d'ions hydronium en quantité notable et que
l'acide phosphorique se comporte comme un monoacide faible de pKa = pKa1.
Le pH de la solution à 0.05 mol.L-1 est de -log0.0156,
soit 1.81.
Si nous employons pour arriver au même résultat
la résolution de l'équation de l'électroneutralité
de la solution d'acide phosphorique.
Nous avons, en application de la démonstration
due aux "nounours", l'équation suivante:
h = (H2A-)
+ 2.(HA2-) + 3.(A3-)
Nous voyons sur le diagramme de prédominance que le
terme relatif à l'anion A3- , anion phosphate ici en
l'occurence, est négligeable dans le cas de l'acide phosphorique
vu que le pKa3 de l'acide est de 12.1. L'anion phosphate est
ultra minoritaire à pH acide, ce qui est le cas ici.
Le terme dû à l'anion HA2-, en
l'occurence à l'anion hydrogénophosphate, est lui aussi
a priori négligeable car le pH supposé d'une solution d'acide
phosphorique à 0.05 mol.L-1 est plus petit que 6.2, c'est à
dire plus petit que la valeur de pKa2-1.
On aura alors à résoudre l'équation
suivante et à vérifier que la solution trouvée est
bien en accord avec les hypothèses que nous venons de formuler.
On arrive à h = (H2A-) ,
c'est à dire à l'équation d'électroneutralité
d'un monoacide de pKa égal à pKa1 ici.
On résout donc une équation du second degré
qui sera:
h2 + Ka1.h -C0.Ka1
= 0
On a C0 qui vaut 0.05 mol.L-1 et Ka1
qui vaut 7.08.10-3.
Soit une valeur de h qui conduit à pH = 1.81.
On voit qu'une telle valeur de pH rend les hypothèses
faites raisonnables et qu'on a eu raison de négliger les ions hydrogénophosphate
et, a fortiori, les ions phosphate.
L'acide phosphorique s'est bien comporté comme
un monoacide faible de pKa égal à pKa1.
Etudions maintenant le cas d'une solution d'acide
citrique, noté H3Ci,de concentration égale à
0.05 mol.L-1.Les pKai respectifs de l'acide citrique
sont: 3.10, 4.80 et 6.40.
La réaction prépondérante est toujours,
comme dans le cas de l'acide phosphorique la réaction correspondant
à l'instauration du premier équilibre chimique, de constante
Ka1 égale à 7.94.10-4, soit 10-3.10.
Comme pour ce qui a été écrit précédemment
on arrive à l'équation du second degré suivante:
x2 + x.7.94.10-4
- 3.97.10-5 = 0
La seule solution physiquement acceptable donne x = 0.0059.
A partir de là envisageons la seconde réaction
prépondérante, celle qui a pour constante d'équilibre
Ka2, de valeur 1.59.10-5.
On arrive à l'équation du second degré
suivante:
x2 + 0.0059.x - 9.3.10-8
= 0
On arrive au résultat suivant: x = 1.57.10-5.
On voit que la quantité d'ions hydronium fournie
par la réaction prépondérante secondaire est négligeable
devant la première, soit quelque chose comme 0.26% du total des
ions hydronium.
On en conclut que l'acide citrique, tout comme l'acide
phosphorique, et malgré des pKai très rapprochés,
se comporte comme un monoacide faible de pKa égal à pKa1.
Le pH de la solution d'acide citrique à 0.05 mol.L-1
est donc de 2.23.
Si l'on utilise la résolution de l'équation
de l'électroneutralité il faut résoudre l'équation
suivante:
h = (H2Ci-) + 2.(HCi2-)
+ 3.(Ci3-)
Pour une solution d'acide citrique de concentration 0.05
mol.L-1 le pH attendu est forcément compris entre 1 et 7. Le diagramme
de prédominance permet a priori d'écarter le terme en citrate
Ci3- mais beaucoup plus difficilement celui en hydrogénocitrate
HCi2-. La résolution du problème par cette méthode
est plus difficile que dans le cas de l'acide phosphorique où les
pKai sont très nettement différents.
Ici on peut tester l'approximation monoacide faible de
pKa égal à pKa1 et vérifier si le résultat
est cohérent avec l'hypothèse faite.
A défaut il faudra résoudre le polynôme
obtenu par l'équation d'électroneutralité.
La méthode de la réaction prépondérante
me paraît, pour le cas de l'acide citrique, nettement plus rentable.
Retour
au plan du cours sur les pH des solutions aqueuses.
II.3.f.Les polybases faibles.
Retour
au plan du cours sur les pH des solutions aqueuses.
On cherchera à calculer le pH d'une solution aqueuse
de carbonate de disodium , communément appelé carbonate de
sodium, de formule Na2CO3 et de
concentration égale à 0.05 mol.L-1
Les deux pKa de l'acide carbonique H2CO3,
ou dioxyde de carbone en solution aqueuse, sont respectivement 6.4 et 10.3.
On appliquera la méthode de la réaction
prépondérante.
Les équilibres acidobasiques qui entrent en jeu
sont les suivants:
CO32- + H2O HCO3-
+ HO-
La constante de cet équilibre est Kb2,
de valeur 2.10-4.
Le second équilibre est:
HCO3- + H2O CO2
+ HO-
La constante de cet équilibre est Kb1,
de valeur 2.5.10-8.
L'application de la méthode de la réaction
prépondérante conduit à l'équation du second
degré suivante:
w2
+ 2.10-4.w -10-5
= 0
La résolution conduit à un pH de 11.49.
On regarde alors si le second équilibre peut être
considéré comme réaction prépondérante
secondaire;
On résout alors l'équation du second degré
suivante:
w2
+ 0.00306.w -7.65.10-11
= 0
On trouve que w vaut 2.4.10-8.
Cette valeur est négligeable devant la valeur
de w trouvée à l'aide de
l'équilibre du début. Le pH de la solution aqueuse de carbonate
de sodium à 0.05 mol.L-1 est donc de 11.49.
Le carbonate de sodium s'est donc comporté comme
une monobase faible de pKb égal à pKb2.
Remarque on pouvait essayer de trouver cette valeur à
l'aide de la formule approchée suivante:
pH = 7 + 0.5.(pKa2 +
logC0).
Formule vraie si le pH trouvé par cette formule est
strictement supérieur à pKa2 + 1.
Ici c'est le cas et on pouvait l'utiliser sans problème.
A défaut on peut résoudre l'équation
de l'électroneutralité.
L'équation d'électroneutralité s'écrit
alors:
h + (Na+) = w
+ (HCO3-) + 2.(CO32-)
Bien entendu ici on a w
qui est plus grand que h, qu'on pourra négliger d'emblée.
Le pH d'une polybase étant forcément basique.
Le pH de la solution ayant, a priori, une valeur supérieure
nettement à 7, on peut considérer que la teneur en
dioxyde de carbone CO2 dissous est négligeable
et que, par conséquent, le carbonate de sodium se comporte comme
une monobase faible de pKb égal à pKb2.
En effet la forme "CO2" ne commence à devenir "importante"
qu'à partir de pH 7.4 (pKa1 + 1). On peut faire l'hypothèse
inverse (à savoir qu'il se comporte comme une dibase) et vérifier
aisément qu'on retombe bien sur la simple et bonne approximation
de la monobase faible avec le résultat obtenu.
Retour
au plan du cours sur les pH des solutions aqueuses.
II.3.g. Les acides aminés.
Retour
au plan du cours sur les pH des solutions aqueuses.
Les acides aminés sont des composants essentiels
de l'organisme puisqu'ils entrent dans la composition des protéines.
Leur formule générale est de la forme:
H2N-CHR-CO2H
Le groupement R est égal :
1. soit à un atome d'hydrogène; ce sera
la "glycine", anciennement appelée "glycocolle".
2. soit à une chaîne hydrocarbonée
ne comportant ni fonction acide ni fonction basique; ce seront les acides
aminés neutres.
3. soit à une chaîne hydrocarbonée
comportant une ou plusieurs fonctions acides; ce seront les acides aminés
"acides".
4. soit à une chaîne hydrocarbonée
comportant une ou plusieurs fonctions basiques; ce seront les acides aminés
"basiques".
Les acides aminés possèdent en général
(sauf pour le cas de la glycine) un atome de carbone asymétrique.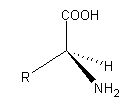
La configuration spatiale des acides aminés naturels
les range dans ce qu'on appelle la série "L".
Au niveau de la nomenclature en stéréochimie
nous verrons en seconde
année que les acides aminés naturels peuvent être,
soit "S", soit "R", suivant la nature du groupement R.
Il existe des acides aminés particuliers, tels
que la proline, qui eux, ont une forme cyclique. Ce sont des cas particuliers:
ce ne sont pas des acides aminés avec une fonction amine "primaire"
mais "secondaire". Le cours de chimie organique de seconde année
viendra éclaircir tout ça.
La forme neutre H2N-CHR-CO2H est
une forme qui est en fait "contredite" par certaines propriétés
physiques, notamment le point de fusion , particulièrement élevé
de ces acides. Leur forte solubilité dans l'eau également
ne plaide pas pour cette forme "neutre".
Il existe un équilibre entre cette forme neutre
et une forme privilégiée, doublement ionisée, qu'on
appellera "amphion" ou "sel interne" ou "zwitterion" (zwitterion est un
mot allemand).
H2N-CHR-CO2H H3N+-CHR-CO2-
Au niveau acidobasique les acides aminés sont en général
le siège de deux équilibres acidobasiques fondamentaux.
Le premier est caractérisé par une constante
d'acidité Ka1:
H3N+-CHR-CO2H
+ H2O H3N+-CHR-CO2-
+ H3O+
Par commodité dactylographique on abrègera
l'écriture de l'équilibre par:
"cation" + H2O "amphion"
+ H3O+
Le second est caractérisé par une constante
d'acidité Ka2:
H3N+-CHR-CO2-
+
H2O H2N-CHR-CO2-
+ H3O+
Par commodité dactylographique on simplifiera l'écriture
par:
"amphion" + H2O "anion"
+ H3O+
Prenons l'exemple d'une solution d'alanine (R = CH3)
de concentration 0.1 mol.L-1 dont on veut connaître le
pH. Les pKai respectifs de l'alanine sont: 2.35 et 9.70.
D'après ce que l'on a dit en début d'exposé
on voit qu'on met en solution aqueuse l'amphion au départ.
Que devient l'amphion?
L'amphion peut avoir a priori une réaction où
il joue le rôle d'un acide au sens de BRONSTED.
"amphion" + H2O "anion"
+ H3O+
La constante de l'équilibre est Ka2 de
valeur 10-9.70, soit 2.10-10.
L'amphion peut avoir aussi une réaction où
il intervient comme une base au sens de BRONSTED.
"amphion" + H2O "cation"
+ HO-
La constante de cet équilibre est Kb1,
de valeur 2.24.10-12, soit 10-11.35.
La question qui se pose est la suivante: le pH est-il
plutôt "acide" ou plutôt "basique"?
Jusqu'à présent on s'est contenté
de faire réagir l'espèce dissoute avec l'eau. Mais, ici?
Ici on a affaire à une espèce qui est à la fois "basique"
et à la fois "acide", donc qui est amphotère.
L'idée est alors la suivante: et si l'ampholyte
réagissait sur lui même? Bref, 1 ampholyte "acide" sur 1 ampholyte
"basique" ça donne quoi?
On écrit alors l'équation suivante:
amphion + amphion cation
+ anion
ou, plus simplement encore:
2 amphion cation
+ anion
La constante Keq de l'équilibre a pour expression
littérale:
Keq = 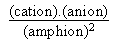 La démostration est incluse dans le cours de thermodynamique
chimique de première année. Le terme au carré vient
de ce que l'on appellera les "potentiels chimiques".
La démostration est incluse dans le cours de thermodynamique
chimique de première année. Le terme au carré vient
de ce que l'on appellera les "potentiels chimiques".
Que vaut la constante Keq de cet équilibre?
Il faut chercher, par tous les moyens licites, à
trouver un "bon" produit de constantes qui donneront ce que l'on cherche.
C'est une partie qui fait un peu "cuisine" mais bon, la cuisine, quand
elle est bonne est-ce vraiment à négliger?
La constante Keq peut se trouver à partir
de: K= Ke.Keq, soit: K = 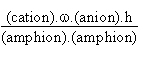
Or on voit que K est le produit de deux autres constantes:
K = Kb1.Ka2.
D'où Keq vaut tout simplement: 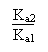
Numériquement, pour l'alanine, vu que les deux
pKa de l'acide aminé valent respectivement 2.35 et 9.70 on trouve
que Keq vaut 4.4.10-8.
A partir de là on voit que Keq est
la constante d'équilibre relative à la réaction
prépondérante. C'est elle qui a la plus grande des valeurs
de toutes les constantes d'équilibre pouvant intervenir dans le
milieu réactionnel, avec possiblilté de transformation "quantitative"
de matière (ici en l'occurence transformation de l'amphion):
Keq est supérieure à Ka2
qui est supérieure à Kb1.
Comment évoluent les quantités de matière
de l'amphion, du cation et de l'anion à partir de la réaction
prépondérante?
On fera un tableau.
Les quantités de matière, exprimées
en mol sont données paur 1 litre de solution.
|
Amphion |
Cation |
Anion |
Instant initial, mise en solution de l'acide
aminé sous la forme d'amphion |
0.1 mol |
0 mol |
0 mol |
| Instant où l'équilibre caractérisé par
Keq est atteint |
0.1-2.x mol |
x mol |
x mol |
On est amené à résoudre une équation
du second degré. Après avoir remplacé par leurs expressions
respectives Keq, (amphion), (cation) et (anion),à
savoir: 4.4.10-8, (0.1-2.x), (x) et (x), on arrive à:
x2 = 4.4.10-8.(0.1-2.x)2.
On remarque de suite , vu que les quantités de matière
sont forcément positives, qu'on peut tout de suite prendre la racine
carrée de l'ensemble. D'une équation du second degré
on se retrouve tout juste avec une équation du premier degré.
On arrive alors tout simplement à la valeur x
= 2.1.10-5.
Cette valeur trouvée pour x imposerait la concentration
en cation et en anion dans le milieu.
A partir de là, si tout restait en l'état,
on pourrait connaître le pH grâce par exemple à la constante
d'acidité Ka1. En effet, on connaît la valeur de Ka1,
la valeur de la concentration en amphion et de celle en cation. La seule
inconnue est h.
On peut procéder de même avec Ka2,
dont on connaît la valeur, de même qu'on connaît la valeur
des concentrations en anion et amphion.
On peut procéder encore plus simplement en faisant
le produit des deux constantes Ka1 et Ka2 et alors
on arrive tout simplement à la formule magique:
pH = 0.5.(pKa1 + pKa2)
Il faut alors vérifier cette hypothèse en recherchant
s'il n'existe pas une réaction prépondérante secondaire,
susceptible de jouer les trublions, et qui produirait une quantité
"notable" d'ions hydronium, voire hydroxyde, afin de modifier le pH .
La réaction prépondérante secondaire
est sans équivoque alors la réaction d'hydrolyse partielle
"acide" de l'amphion en anion et ion hydronium, bref la réaction
caractérisée par la constante d'équilibre Ka2.
Cette constante qui vaut numériquement ici 10-9.70,
soit 2.10-10, est en effet supérieure à Kb1,
de valeur 2.24.10-12 relative, quant à elle, à
l'hydrolyse partielle "basique" de l'amphion.
Ces valeurs sont agrémentées du fait nécessaire
mais non suffisant qui consiste à bien mettre en avant qu'il y a
suffisamment de quantité de matière, en l'occurence suffisamment
de moles d'amphion, susceptibles d'être transformées soit
selon l'hydrolyse acide partielle, soit selon l'hydrolyse basique partielle.
On arrive alors au tableau suivant, exprimé en
mole de substance pour 1 litre de solution. Les ions de l'eau seront comptés
pour négligeables dans le bilan de matière, vu que la quantité
de matière de l'anion est au départ sensiblement 210 fois
plus grande que celle des ions de l'eau, notamment l'ion hydronium:
|
Amphion |
Anion |
Ion hydronium (oxonium) |
| Instant initial, une fois la réaction prépondérante
instaurée |
0.1 - 4.2.10-5 mol, soit:
0.1 mol |
2.1.10-5 mol |
0 mol |
| Instant final, une fois la réaction prépondérante
secondaire instaurée |
0.1 -x mol |
2.1.10-5 +x mol |
x mol |
On arrive alors devant l'équation du second degré
suivante:
2.10-10 =  On résout alors l'équation:
x2 + 2.10-5.x -2.10-11
= 0
On arrive à trouver x = 9.12.10-7.
On résout alors l'équation:
x2 + 2.10-5.x -2.10-11
= 0
On arrive à trouver x = 9.12.10-7.
On en déduit que le pH de la solution vaut alors
-log(9.12.10-7), soit 6.04.
Remarque: si l'on avait appliqué la formule "magique"
pH = 0.5.(pKa1 + pKa2), on trouvait la même
valeur. Je préfère considérer cette formule comme
purement "mnémotechnique" plutôt que de la prendre pour un
raisonnement.
Le problème de cette formule "magique" c'est qu'elle
ne marche pas toujours. Notamment lorsque la concentration des acides aminés
est trop faible.
L'exemple en sera fourni par le calcul du pH d'une solution
aqueuse d'alanine de concentration égale cette fois à 0.01
mol.L-1.
Le calcul du rendement en quantité de matière
à partir de la réaction prépondérante donne,
de façon analogue à l'exemple précédent, la
valeur de 2.1.10-6 mol de cation et 2.1.10-6 mol
d'anion à partir de 0.01 mol d'amphion.
La réaction prépondérante secondaire,
à savoir l'hydrolyse partielle acide de l'amphion, de constante
d'équilibre Ka2 conduit à la résolution
de l'équation du second degré suivante:
Ka2 = 10-9.70 = 2.10-10
= 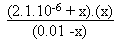 Soit, de façon traditionnelle:
x2 + 2.1.10-6.x
-2.10-12 = 0
La résolution conduit à x = 7.11.10-7.
Soit, de façon traditionnelle:
x2 + 2.1.10-6.x
-2.10-12 = 0
La résolution conduit à x = 7.11.10-7.
Le pH cherché est de 6.15.
Je me suis amusé à résoudre le problème
en tenant compte des 10-7 mol d'ions hydronium présents
par litre de solution dès le départ. Tous calculs faits je
trouve un pH de...6.14, bref, la même chose.
Dans les calculs sur les pH des solutions d'acides aminés
on demande parfois le pH des chlorhydrates d'acides aminés.
Qu'est ce qu'un chlorhydrate?
On obtient un "chlorhydrate" en faisant réagir
mole à mole un acide aminé, donc un amphion, avec l'acide
chlorhydrique.
Je prends l'exemple de l'alanine. L'équation chimique
est la suivante:
H3N+-CHCH3-CO2-
+ H3O+ H3N+-CHCH3-CO2H
+ H2O
H3N+-CHCH3-CO2H
+ H2O
On obtient ainsi une forme souvent nettement plus soluble
dans l'eau que la forme amphionique.
Le composé obtenu s'appelait "autrefois" le chlorhydrate
d'alanine, aujourd'hui on l'appelle "chlorure d'alanine-ammonium".
Le pH de la solution de chlorhydrate fait appel à
la réaction prépondérante.
La connaissance des deux pKai de l'alanine
montre que c'est la réaction caractérisée par une
constante d'équilibre égale à Ka1 qui est
la réaction prépondérante. En effet Ka1
est très nettement supérieur à Ka2 ici.
On se propose de calculer le pH d'une solution à
0.05 mol.L-1 de chlorhydrate d'alanine.
Le chlorhydrate se comporte comme un monoacide faible
de pKa égal à pKa1. On arrive alors à une
équation du second degré:
x2 + 4.47.10-3.x
- 2.24.10-4 = 0
La résolution mène à un pH de 1.89.
On pose aussi souvent la question du pH d'une solution
d'alaninate de sodium, à savoir une solution de la forme anionique
de l'acide aminé.
L'équation de formation de l'alaninate (pas facile
à dire!!!) de sodium à partir de l'alanine est la suivante:
H3N+-CHCH3-CO2-
+
Na+ + HO- H2N-CHCH3-CO2-
+ Na+ + H2O
Le pH d'une solution d'alaninate de sodium à 0.05
mol.L-1 fera appel là aussi à la réaction
prépondérante.
L'exament des diverses constantes d'équilibre
susceptibles d'être les réactions prépondérantes
amène à choisir la réaction caractérisée
par la constante Kb2 comme réaction prépondérante.
On a en effet Kb2 qui est très nettement supérieure
à Kb1: 10-4.3 pour Kb2 contre 10-11.65
pour Kb1.
L'alaninate de sodium se comporte comme une monobase
faible de pKb égal à pKb2.
On arrive alors à une équation du second
degré en w qui est la suivante:
w2
+ 5.10-5.w -2.5.10-6
= 0
La résolution nous conduit aisément à
un pH de 11.19.
Retour
au plan du cours sur les pH des solutions aqueuses.
Suite du
cours de chimie sur les pH des solutions aqueuses. Cliquer ici.