Retour sommaire des TP de chimie générale.
I. Principe de la conductimétrie.
On se reportera au contenu introductif de la manipulation
8, relative à l' étude de la saponification
de l' éthanoate d' éthyle par l' hydroxyde de sodium, cinétique
d' ordre 2, suivie par conductimétrie.
II. Conductivité des électrolytes.
Afin de pouvoir comparer la conductivité des différents électrolytes entre eux on définit la conductivité équivalente L par:
La conductivité des électrolytes varie avec la concentration.
On définit alors la "conductivité équivalente limite" L 0, conductivité équivalente de l' électrolyte lorsque sa concentration tend vers 0, valeur bien entendu extrapolée.
Dans le cas des électrolytes dits "forts", tels
que le chlorure de sodium NaCl, l' acide chlorhydrique H3O++
Cl-, l' éthanoate de sodium NaCH3CO2,
il y a une relation linéaire, dite loi de KOHLRAUSCH, entre L
et c 0.5.
L' extrapolation de chacune de ces droites pour c
0.5
= 0 donnera dans chaque cas la valeur de L0.
Dans le cas des électrolytes dits "faibles", tels que l' acide éthanoïque CH3CO2H, cette linéarité n' existe plus. Lorsque la concentration c de l' électrolyte tend vers 0 alors L tend vers l' infini.
On calculera alors L 0 en utilisant la loi de KOHLRAUSCH qui s' énonce par:
La conductivité d' un électrolyte est la somme des conductivités propres, indépendantes, de chacun de ses ions.
On aura alors, étant donné que l' acide éthanoïque CH3CO2H découle formellement de la réaction suivante:
Na+ + CH3CO2- +
H3O+ + Cl- ![]() CH3CO2H
+ Na+ + Cl- + H2O
CH3CO2H
+ Na+ + Cl- + H2O
1. Pour chaque constituant:
2. Pour obtenir L0 CH3CO2H:
III. Détermination du degré de dissociation a de l' acide éthanoïque.
On utilisera la relation:
a = 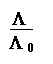
On rappelle que pour un acide faible HA, tel que l' acide éthanoïque CH3CO2H, on a la relation suivante entre Ka, constante d' acidité, a,degré de dissociation, et C concentration de l' acide:
K a = 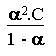
La concentration C est exprimée en mol.L-1.
IV. Manipulation.
Pour chacun des quatre électrolytes de la manipulation,
à savoir l' acide chlorhydrique, le chlorure de sodium, l' acide
éthanoïque et l' éthanoate de sodium, préparer
une concentration C initiale de chacun d' eux égale à 0.1
mol.L-1. Le volume de solution préparée sera choisi
égal, par exemple, à 200 mL.
1. Pour chacun des quatre électrolytes faire les dilutions nécessaires de façon à préparer au moins 100 mL de solutions aux concentrations suivantes: 0.05 mol.L-1, 0.025 mol.L-1 et 0.0125 mol.L-1.
2. Pour chacune des cinq concentrations d' un électrolyte donné lire la valeur de la conductivité s correspondante.
3. Pour chacune des cinq concentrations d' un électrolyte donné calculer L.
4. Tracer sur une feuille de papier millimétré,
pour chacun des trois électrolytes forts, à savoir le chlorure
de sodium, l' éthanoate de sodium et l' acide chlorhydrique, la
représentation linéaire L
= f ( c 0.5).
En déduire la valeur extrapolée à
0 de chacune des trois conductivités équivalentes limites
L
0 correspondantes.
5. Calcul de L0 pour l' acide éthanoïque à partir des trois valeurs précédentes, en application de la loi de KOHLRAUSCH.
5. Calcul du degré de dissociation a pour chacune des cinq concentrations. Vérification de la relation liant la constante d' acidité Ka à la concentration C et au degré de dissociation a.
6. Calcul du pKa du couple CH3CO2H / CH3CO2-. Comparer avec la valeur théorique à 25°C, égale à 4.75.
Retour au sommaire des TP de Chimie générale.
Retour au sommaire des TP de chimie générale.
I. Chromatographie sur papier. Recherche de l' acide malique dans le vin et les jus de fruits.
L' acide malique, ou acide 2 hydroxy butane dioïque,
HO2C-CHOH-CH2-CO2H, est un constituant
de certains fruits, notamment la pomme, d' où il tire son nom en
latin, malus, mais aussi le raisin.
Il est biologiquement instable et de nombreux microorganismes,
tels que les bactéries lactiques, peuvent le métaboliser
en acide lactique, ou acide 2 hydroxy propanoïque, HO2C-CHOH-CH3.
Dans les vins en cours d' élaboration le passage de l' isomère S (-) de l' acide malique (isomère seul présent naturellement dans le raisin) à l' isomère S (+) de l' acide lactique, avec dégagement de dioxyde de carbone CO2, constitue ce que l' on appelle la fermentation malolactique:
HO2C-CHOH-CH2-CO2H ![]() CO2 + HO2C-CHOH-CH3
CO2 + HO2C-CHOH-CH3
La stéréochimie de la réaction est donc spécifique et se produit avec rétention de la configuration du carbone asymétrique de la molécule d' acide malique.
La fermentation malolactique intervient après la fermentation alcoolique et agit comme agent de stabilisation biologique du vin, en même temps qu' elle induit des arômes de fermentation particuliers, notamment avec des dérivés tels que le diacétyle, ou butane dione CH3-CO-CO-CH3, le butan 2,3-diol, CH3-CHOH-CHOH-CH3, ou l' acétoïne, alias butan 2-one, 3-ol, CH3-CHOH-CO-CH3.
I.1. Principe.
La chromatographie sur papier d' un dépot de vin
permet de séparer les acides organiques de ce dernier.
Après révélation des tâches
on observera ainsi trois spots d' acides au maximum:
1. L' acide tartrique, HO2C-CHOH-CHOH-CO2H,
à la partie inférieure.
2. L' acide lactique, CH3-CHOH-CO2H,
non séparé de l' acide succinique, HO2C-CH2-CH2-CO2H,
à la partie supérieure.
3. L' acide malique, éventuellement présent,
au centre.
Par comparaison avec des solutions étalon on peut estimer la teneur en acide malique dans le vin.
I.2. Mode opératoire.
1. Le solvant révélateur est placé
dans la cuve de verre.
Il est constitué par un mélange de 40 mL
de butan 1-ol et de 20 mL d' acide éthanoïque à 50 %,
auxquels on aura ajouté environ 10 mg de bleu de bromophénol.
2. A environ 3 cm du bas de la feuille de papier à chromatographie tracer au crayon à papier une ligne horizontale. A intervalles réguliers, marquer les futurs emplacements des dépots par une croix et un numéro.
3. Déposer avec une pipette Pasteur, sur chaque marque effectuée précédemment, un dépot de l' ordre de grandeur de 10 mL de chaque échantillon à analyser.
4. Déposer également une goutte de chaque solution étalon d' acide malique à 5 g.L-1, 2.5 g.L-1 et 1.25 g.L-1.
5. Agrafer les bords de la feuille de papier sans qu' il y ait de chevauchement des deux bords. Déposer la feuille ainsi agrafée dans la cuve. Couvrir, afin que le système soit un système thermodynamiquement "fermé" et dont la composition du mélange éluant soit constante.
6. Attendre jusqu' à ce que le mélange éluant soit monté jusqu' à environ 2 cm du bord supérieur de la feuille de papier. Cela peut prendre 2 h.
7. Révéler ensuite la feuille en la séchant au sèche-cheveux . La feuille prend une coloration bleue et les spots d' acide restent jaunes. Veiller à ne pas mettre les doigts sur la feuille.....
8. Déterminer le rapport frontal Rf de l' acide malique dans les conditions opératoires données et conclure quant à sa teneur, éventuelle, dans les différents vins et jus de fruits étudiés.
II. Chromatographie sur colonne remplie. Séparation de pigments naturels, carotènes et chlorophylles.
Ancienne manip de la MST Mer à Toulon (avant 1992).
On se propose de séparer des pigments colorés contenus dans une plante par la technique de la chromatographie liquide sur colonne remplie.
II. 1. Manipulation. Préparation de l' extrait.
1. Broyer environ 1 gramme d' herbe ou de tout autre végétal
dans environ 5 mL d' acétone pure.
2. Séparer les phases liquide et solide par transvasement.
3. Recommencer l' opération trois fois.
4. Filtrer sur tampon en laine de verre les trois extraits
recueillis, afin d' éliminer les particules solides.
5. Ajouter 2 mL de 1,4 dioxanne, homogénéiser,
ajouter ( en agitant la solution) goutte à goutte, en utilisant
une pipette Pasteur, de l' eau distillée jusqu' à formation
d' un précipité.
Cette opération permet de séparer les composés
xanthophylles de la chlorophylle A, de la chlorophylle B, des carotènes
contenus dans le précipité.
6. Centrifuger. Attention à l' équilibre
de la centrifugeuse lors de l' opération de centrifugation!!!!!
Utiliser impérativement la balnce pour bien équilibrer les
deux tubes.
7. Redissoudre le culot de centrifugation, après
avoir éliminé le liquide surnageant, dans un minimum d' acétone,
c' est à dire dans environ 1 à 3 mL.
8. Ajouter à cette solution un volume égal
d' éther de pétrole.
9. Placer l' ensemble de cette solution dans une petite
ampoule à décanter contenant une solution de chlorure de
sodium à 10%.
10. Effectuer trois extractions en évacuant
l' eau salée qui contient les composés hydrphiles et l' acétone.
11. Sécher la phase organique avec un minimum
de sulfate de sodium anhydre.
La phase organique séchée est ensuite transférée
en tête de colonne de chromatographie.
II.2. Séparation et purification.
II.2.1. Obtention de la colonne par la méthode dite de la colonne humide. Elution fractionnée des divers constituants.
1. Remplir la colonne de chromatographie avec de l' éther
de pétrole, après l' avoir munie à sa base d' un petit
coton en laine de verre si jamais on ne dispose pas de colonne de chromatographie
munie d' un verre fritté .
2. Ajouter lentement, en pluie, la poudre de cellulose
afin de remplir lentement, par décantation, la colonne aux deux-tiers.
3. Faire vibrer légèrement la colonne afin
de parfaire le tassement. Ajouter, si besoin, de la poudre de cellulose.
4. Placer un petit tampon de laine de verre en tête
de colonne et procéder, à l' aide d' une tige de verre, au
tassement de la poudre de cellulose.
Le petit coton en laine de verre permet de limiter les
remous lors des additions de solvant.
5. Ajuster, par écoulement du liquide, le niveau
du mélange éluant (le solvant d' élution) avec le
sommet du coton en laine de verre.
6. Introduire l' extrait et faire adsorber ce dernier
par la colonne de cellulose.
Ceci a pour but d' éviter la remise en solution
dans le mélange éluant de l' ensemble de l' extrait.
7. Eluer par de l' éther de pétrole jusqu'
à séparation des carotènes, de couleur jaune bouton
d' or.
8. Collecter les carotènes dans des tubes à
essais afin de pouvoir les analyser ultérieurement par spectrophotométrie
UV-visible.
9. Procéder ensuite à une élution
avec des solutions éther de pétrole-acétone de polarité
croissante.
On commencera par un mélange volume/volume éther
de pétrole-acétone 96-4, afin de collecter les fractions
xanthophylles, puis ensuite par un mélange 94-6 afin de collecter
la chlorophylle A, enfin par un mélange 92-8 afin de collecter la
chlorophylle B.
II.2.2. Spectres d' absorption UV-visible des différentes fractions.
1. Enregistrer la ligne de base de l' appareil en utilisant
si possible des cuves en quartz (attention!!! CHER!!!) transparentes aux
UV.
2. Enregistrer les spectres d' absorption des diverses
fractions.
3. Interpréter les spectres en référence
avec les spectres des composés purs.
| Type de pigment. | Maximums d' absorption en nanomètres (dans les solvants organiques). | Espèces contenant les pigments. |
| Chlorophylles | . | . |
| Chlorophylle a | 420, 660 | Toutes les plantes supérieures et les algues. |
| Chlorophylle b | 435, 643 | Toutes les plantes supérieures et les algues vertes. |
| Chlorophylle c | 445, 625 | Diatomées et algues brunes. |
| Chlorophylle d | 450, 690 | Algues rouges. |
| Caroténoïdes | . | . |
| b carotène | 425, 450, 480 | Plantes supérieures et la plupart des algues. |
| a carotène | 420, 440, 490 | La plupart des plantes et quelques algues. |
| Lutéol | 425, 445, 475 | Algues vertes, rouges et plantes supérieures. |
| Violaxanthol | 425, 450, 475 | Plantes supérieures. |
| Fucoxanthol | 425, 450, 475 | Diatomées et algues brunes. |
| Phycobilines | . | . |
| Phycoérythrines | 490, 546, 576 | Algues rouges, certaines algues bleu-vert. |
| Phycocyanines | 618 | Algues bleu-vert et certaines algues rouges. |
Retour sommaire des TP de Chimie générale.
Manipulation 19: détermination par spectrophotométrie visible du pKa d' un indicateur coloré, le BBT.
Retour au sommaire des TP de chimie générale.
I. Introduction.
On envisage le cas très classique du bleu de bromothymol,
le BBT, indicateur coloré de fin de réaction entre un acide
fort et une base forte.
La teinte de cet indicateur varie du jaune, à
pH acide au bleu, à pH basique, la teinte sensible se situant
autour de pH 7.
On préparera une solution aqueuse de BBT de l' ordre de 2.10-3 mol.L-1.
Bien entendu la manipulation peut être reprise avec n' importe quel autre indicateur coloré dont les trois teintes sont situées dans le domaine du spectre visible.
II. Mesures.
II.1. Spectre en milieu acide, à pH 1.
La solution est faite avec 5 mL de la solution mère d' indicateur, 10 mL d' acide chlorhydrique à 1 mol.L-1, et 85 mL d' eau distillée.
1. Enregistrer le spectre d' absorption dans le visible
(400-800 nm) de la solution constituée, après avoir fait
le zéro optique de l' appareil.
2. Relever le maximum d' absorption et noter la valeur
l
1
max correspondant à la longueur d' onde de ce maximum, ainsi
que la valeur de la densité optique DO 1 à cette
longueur d' onde.
II.2. Spectre en milieu basique, à pH 13.
La solution est faite avec 5 mL de la solution mère d' indicateur, 10 mL de soude à 1 mol.L-1, et 85 mL d' eau distillée.
1. Enregistrer le spectre d' absorption dans le visible
de la solution.
2. Noter la valeur de
l 2 max, ainsi que la valeur
de la densité optique DO 2, à cette longueur d'
onde.
II.3. Spectre dans la zone sensible de l' indicateur coloré, entre pH 6 et 7.6.
1. Préparer une solution tampon à l' aide du tampon dihydrogénophosphate / hydrogénophosphate H2PO4- /HPO42-, dont le pKa vaut 7.2 à 25°C.
La solution à étudier est faite avec 5 mL de solution mère de BBT, 10 mL de solution tampon et 85 mL d' eau distillée.
2. Enregistrer le spectre dans le visible de la solution
obtenue.
Noter la présence de deux maximums d' absorption
, l' un pour l 1
max, l' autre pour l2
max, et noter également les valeurs correspondantes des densités
optiques DO' 1 et DO' 2.
Noter la présence d' un point singulier, les trois
courbes DO = f ( l
) se coupant en un même point, le point isobestique.
Noter que la longueur d' onde du point isobestique est
proche de 500 nm pour le BBT.
III. Calcul du pKa de l' indicateur coloré.
On se reportera, au niveau des définitions, à mon cours sur les indicateurs colorés.
On pose:
1. a = (HIn), à savoir concentration de la forme
acide HIn de l' indicateur coloré.
2. b = (In-), à savoir concentration
de la forme basique In- de l' indicateur coloré.
3. c = a + b = c = (HIn) + (In-) , à
savoir concentration globale de l' indicateur.
Noter bien que c est une constante ici.
Plaçons-nous à l 1 max.
On pose:
e HIn (l 1 max) et e In- (l 1 max) , les deux coefficients molaires d' extinction, àl1 max, des espèces respectives HIn et In-.
En application de la loi de BEER-LAMBERT, on peut alors écrire, quel que soit le pH du milieu:
DO 1 = e HIn (l 1 max) .a.l + e In- (l 1 max).b.l
avec l qui désigne la longueur du trajet optique du faisceau lumineux.
Si on se situe à pH très acide, la forme HIn est largement majoritaire devant la forme In-.
On en déduit alors, à pH très acide, l' expression approchée de DO 1:
DO 1 = e HIn (l 1 max).c.l
On tirera e HIn (l 1 max) de l' expression précédente.
Si on se situe à pH très basique, alors la forme In- devient largement majoritaire devant la forme HIn et l' on pourra écrire, à l 1 max :
do 1 = e In- (l 1 max).c.l
On tirera e In- (l 1 max) de l' expression précédente.
Si l' on se situe dans la zone sensible, en remplaçant e HIn (l 1 max) et e In- (l 1 max) par leur expression on arrive à:
DO' 1 = e HIn (l 1 max) .a.l + e In- (l 1 max).b.l = (DO 1 .a + do 1.b)/c
On en tire:
c.DO' 1 = a.DO 1 + b.do 1
Or, comme a + b = c, il vient:
(DO 1 - DO' 1).a = (DO' 1 - do 1).b
D' où l' on peut exprimer le rapport b/a:
b/a = (DO 1 - DO' 1) / (DO' 1 - do 1)
D' où l' on tire:
pKa = pH + log (DO 1 - DO' 1) / (DO' 1 - do 1)
Le choix du BBT pour la détermination du point d' équivalence acidobasique lors du dosage d' un acide fort par une base forte vous paraît-il judicieux?
Questions subsidiaires:
1. Rechercher dans la littérature les formules développées des formes HIn et In- du BBT.
2. Conclure quant à la possibilité du BBT d' absorber de la lumière visible.
3. Déterminer, par la même méthode, le pKa du vert de bromocrésol.