Retour au sommaire des TP de chimie générale.
C' est une manipulation de niveau Licence de Chimie. Avec tous mes remerciements à l' Université de Brest.
I. Introduction.
Cette manipulation a pour but d' étudier la cinétique
d' iodation de l' acétone en milieu tampon, ainsi que de déterminer
les ordres partiels de réaction vis-à-vis du diiode I2
et de l' acétone CH3COCH3.
La réaction est catalysée par les acides
et les bases, au sens général de BRONSTED, c' est à
dire, non seulement par les ions H3O+ et HO-,
mais aussi par tous les acides du type HA et leur base conjuguée
A-.
Cette catalyse acidobasique généralisée
sera examinée en opérant en milieu tampon phosphate (couple
dihydrogénophosphate /monohydrogénophosphate) H2PO4-/HPO42-.
Les différentes cinétiques seront suivies
par spectrophotométrie dans le visible. L' évolution de la
densité optique des divers milieux réactionnels en fonction
du temps permettra de déduire les ordres partiels et les constantes
de vitesse.
II. Equations cinétiques.
Soit une réaction:
Les termes a et b
désignent respectivement les ordres partiels de réaction
par rapport aux réactifs A et B.
Néanmoins, on constate, sous l' effet de l' addition
d' un acide ou d' une base, une variation de la valeur de la constante
de vitesse k selon la relation:
Ceci ne peut s' expliquer qu' en admettant une catalyse
par les ions H3O+ ou HO-.
Ceux-ci, en effet, n' étant pas consommés
au cours de la réaction, leur concentration reste constante, bien
qu' ils ne soient pas nécessairement en excès. Par conséquent,
l' ordre apparent devient, par dégénérescence, égal
à a + b.
Le terme en H3O+ ou celui en HO-
entre dans la constante apparente k, qui dépend ainsi de l' acidité.
De façon plus générale k peut être
fonction de toutes les espèces acidobasiques présentes dans
le milieu: c' est cela la catalyse acidobasique généralisée,
traduite par l' équation complète suivante:
Pour mettre en évidence une catalyse acidobasique
généralisée il suffit d' effectuer la réaction
dans un milieu tampon HA /A-. Le rapport des concentrations
(HA) et (A-) est maintenu constant, ce qui entraîne une
valeur de pH pratiquement constante.
On fait alors tout simplement varier (HA) et (A-).
Si la vitesse reste inchangée la catalyse est
spécifique.
Si, au contraire, k est fonction de la concentration
du tampon, alors la catalyse est généralisée.
C. Etude de la réaction catalysée.
C.1. Mécanisme de la réaction.
D' après de nombreuses constatations expérimentales l' iodation (monoiodation) de l' acétone doit s' effectuer en deux stades.
Premier stade: énolisation de l' acétone suivant:
CH3COCH3 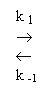 H3C-COH=CH2
H3C-COH=CH2
H3C-COH=CH2
+ I2![]() H3C-CO-CH2I + H3O+ + I-
H3C-CO-CH2I + H3O+ + I-
On peut appliquer l' hypothèse de l' état quasi-sationnaire au composé intermédiaire, à savoir l' énol, c' est à dire qu' on suppose que sa concentration variera très peu au cours du processus global.
Donc:
 = 0
= 0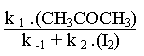
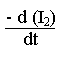 =
k 2 .(I2).(énol)=
=
k 2 .(I2).(énol)= 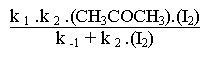
= k
1
.(CH3COCH3)
C.2. Catalyse.
L' étape d' énolisation de l' acétone
est catalysée par les ions H3O+ et/ou les
ions HO-.
La constante de vitesse k 1 et, par conséquent,
l' étape limitante, dépendent non seulement du pH mais aussi
des formes acides et basiques des diverses espèces en présence.
Pour expliquer leur action on a proposé les schémas
réactionnels ci-dessous.
Chacun se décompose en deux étapes.
C.2.a. Cas d' une catalyse acide.
1. Equilibre rapide de protonation de l' acétone par l' acide HA:
 [H3C-COH-CH3]+ + A-
[H3C-COH-CH3]+ + A-C.2.b. Cas d' une catalyse basique:
1. Formation lente de l' énolate:
H3C-CO-CH3 + A- [H3C-CO=CH2]-
+ HA
[H3C-CO=CH2]-
+ HA
[H3C-CO=CH2]- + HA  H3C-COH=CH2
+ A-
H3C-COH=CH2
+ A-
D. Choix du tampon.
Le système équimolaire H2PO4-
/HPO42-
fournit un milieu pratiquement neutre: le pKa du couple est voisin
de 7.2 à 25°C.
On admettra, étant donné également
les deux autres pKa de l' acide phosphorique (2.15 et 12.10 à 25°C)
, que les contributions catalytiques de H3O+ et de
H2PO4- sont négligeables ici, tout
comme celles des formes H3PO4 et PO43-.
Par suite, l' expression de k 1 se réduit
alors à:
E. Vérification expérimentale.
Elle consiste à établir les ordres partiels
par rapport à l' acétone et au diiode, ainsi qu' à
déterminer la valeur de k 1.
E.1. Principes de mesure.
La solution de diiode I2 est préparée dans l' iodure de potassium KI. L' espèce prépondérante est l' anion triiodure I3-, formé selon la réaction équilibrée suivante:
La constante d' équilibre est de l' ordre de 700.
On pourra considérer alors, en application de la loi d' action de
masse, que l' éqilibre est très déplacé vers
la droite, donc vers la formation de l' anion triiodure.
On suivra l' évolution de la concentration de
cette espèce par spectrophotométrie dans le visible, étant
donné son fort caractère coloré.
Comme on suppose a priori que la réaction est d' ordre 0 par rapport au diiode et d' ordre 1 par rapport à l' acétone on doit avoir alors:
 k 1 .(CH3COCH3)
= k observé = constante.
k 1 .(CH3COCH3)
= k observé = constante.D' après la loi de BEER-LAMBERT la densité optique DO est proportionnelle à la concentration de l' espèce absorbante, soit:
D' où:
C' est une droite dont la pente conduit à k observé.
Remarque:
Les essais ont été réalisés
à force ionique I constante, de manière à éviter
toute modification des coefficients d' activité. La valeur du pH
risquerait dès lors de se trouver changée.
E.2. Manipulations.
E.2.1. Mesure de la densité optique DO des solutions.
Enregistrer le spectre de I3- dans le visible. Déterminer arbitrairement la valeur de la longueur d' onde l à laquelle l' ensemble de toutes les mesures seront faites. Connaissant alors (I3-) en déduire la valeur du coefficient d' exctinction molaire e I3- (l). Cette valeur servira bien évidemment pour tous les calculs qui vont suivre.
E.2.2. Solutions de départ.
E.2.2.1. Le mélange tampon. Solution S 1.
Le tampon est un mélange équimolaire de monohydrogénophosphate de disodium Na2HPO4 et de dihydrogénophosphate de sodium NaH2PO4. Les deux concentrations seront prises égales à 0.05 mol.L-1chacune.
E.2.2.2. Solution d' acétone. Solution S 2.
La solution d' acétone à 2 mol.L-1 est préparée en milieu tampon: 0.05 mol.L-1 en chacune des espèces phosphatées précédentes.
E.2.2.3. Solution de lugol (diiode dans de l' iodure de potassium). Solution S 3.
On dissout une quantité convenable de diiode I2 en paillettes dans de l' iodure de potassium KI à 0.2 mol.L-1. La concentration en anion triiodure doit être voisine de 2.10-3 mol.L-1. Etant donné la valeur de la constante d' équilibre K ( valeur voisine de 700 , à 25°C) de la réaction de formation du triiodure de potassium on peut considérer que tout le diiode est consommé par la réaction afin de conduire au triiodure de potassium.
E.2.2.4. Solution de chlorure de sodium. Solution S 4.
On préparera une solution de chlorure de sodium NaCl à 0.2 mol.L-1 afin qu' elle assure le complément éventuel en ions afin de maintenir constante la force ionique I du mélange réactionnel.
E.2.3. Force ionique I.
La force ionique I d' une solution étant égale à:
Avec C i qui désigne la concentration
de l' espèce i, et z i la charge électrique de
l' espèce i.
On montrera que si l' on mélange des volumes V
1,
V 2, V 3 et V 4, quelqconques, avec leur
somme égale à une constante V, de chacune des quatre solutions
précédentes, S 1, S 2, S 3
et S 4, alors on a toujours:
E.2.4. Expériences.
On dispose de toute une batterie de fioles jaugées,
notamment de fioles de 25 mL.
Pour montrer l' influence des trois facteurs
on préparera une série de cinq ou six fioles à chaque fois, pour chaque facteur.
Déterminer LIBREMENT les valeurs des volumes V 1, V 2, V 3 et V 4 des quatre solutions S 1, S 2, S 3 et S 4, afin que l' influence de ces trois facteurs puisse être correctement suivie.
On prendra comme somme des quatre volumes librement choisis, étant donné le matériel de l' université, la valeur de 25 mL.
On complètera un tableau de mesures pour chaque
paramètre étudié séparément, les deux
autres paramètres restant bien entendu fixes.
| . | Fiole 1 | Fiole 2 | Fiole 3 | Fiole 4 | Fiole 5 | Fiole 6 |
| Volume de S 1 | . | . | . | . | . | . |
| Volume de S 2 | . | . | . | . | . | . |
| Volume de S 3. | . | . | . | . | . | . |
| Volume de S4. | . | . | . | . | . | . |
| Volume total (25 mL) | . | . | . | . | . | . |
ATTENTION!!!!!
Le volume de solution de triodure de potassium sera ajouté
au tout dernier moment, lorsqu' on déclenchera le chronomètre.
On prendra une mesure de densité optique toutes
les minutes.
On fera dix mesures par série.
E.3. Interprétation des résultats.
On présentera sous forme de droites les résultats obtenus qui permettent de vérifier l' influence successive des trois facteurs.
On en déduira l' ordre 0 par rapport au diiode
I2, ainsi que l' ordre 1 par rapport à l' acétone
CH3COCH3.
On en déduira l' influence du tampon. On calculera
k HPO42-, ainsi que la valeur de k 1.
Retour sommaire des TP de Chimie générale.
Manipulation 16: cinétique: réduction de l' anion hexacyanoferrate (III), anciennement appelé "ferricyanure", par l' acide ascorbique. Théorie du complexe activé.
Retour sommaire des TP de Chimie générale.
Manipulation de cinétique niveau Licence. Avec l' aimable et précieux concours de l' Université de Brest.
A. Introduction.
Cette manipulation illustre la théorie du complexe
activé, par l' influence de la force ionique I sur la vitesse de
réduction de l' anion hexacyanoferrate (III) (anion ferricyanure)
en hexacyanoferrate (II) (anion ferrocyanure) par l' acide ascorbique,
alias vitamine C.
L' étape limitante de la réaction sera
déterminée.
La réaction sera suivie par potentiométrie,
en tenant compte de la force ionique des solutions, ce qui modifie les
valeurs des potentiels redox.
B. Généralités.
D' un point de vue cinétique, les réactions
réelles sont presque toujours complexes quant à leur mécanisme
et sont, par conséquent, formées de plusieurs étapes
élémentaires.
Dans les cas simples l' une d' elles est plus lente et
constitue l' échelon cinétique limitant.
Une réaction élémentaire, généralement
bimoléculaire, telle que:
A + B ![]() Produits
Produits
peut s' interpréter par la théorie des états
de transition, ou du "complexe activé".
La rencontre de A et de B se traduit par la formation
d' un complexe activé AB* correspondant à un maximum d' énergie
potentielle.
Ce niveau implique que les réactifs franchissent
une barrière de potentiel égale à la différence
d' énergie entre celle qui leur est propre et celle de AB*.
Cette barrière constitue ce que l' on appelle
l' énergie d' activation.
AB* étant supposé être en équilibre
avec ses constituants, on a:
A + B ![]() AB*
AB* ![]() Produits
Produits
L' étape limitante est celle qui correspond
à la disparition du complexe activé.
Dans ces conditions, la vitesse de réaction est
proportionnelle à la concentration du complexe activé:
v = Constante.(AB*)
La vitesse est aussi égale au nombre de molécules
de complexe activé AB* franchissant, par seconde, la barrière
d' activation.
Le facteur de proportionnalité est la fréquence
universelle n:

Avec k B qui désigne la constante de
BOLTZMANN, T la température kelvin et h la constante de PLANCK.
D' où l' on tire:
.(AB*)C.1. Solutions idéales.
La théorie du complexe activé établie
à partir de réactions en phase gazeuse concernant des gaz
parfaits, peut s' appliquer également aux solutions idéales.
Soit K C* la constante relative aux concentrations:
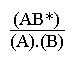 . K C* . (A).(B)
. K C* . (A).(B)L' étude cinétique d' une réaction bimoléculaire du type:
A + B ![]() Produits
Produits
a pour expression:
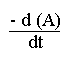 = k 0 .(A).(B).K
C*
= k 0 .(A).(B).K
C*C.2. Solutions ioniques réelles.
Soient les espèces A et B, de charges respectives z A et z B. La réaction élémentaire avec formation intermédiaire d' un complexe activé de charge z A + z B s' écrit alors:
A z A + B z
B![]() (AB*)
z
A
+ z B
(AB*)
z
A
+ z B![]() Produits
Produits
A partir de là, en tenant compte des facteurs d' activité:
K C* = 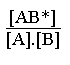 .
. 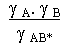
D' où l' on tire:
. K C* .
.[A].[B] = k expérimentale .[A].[B]
. K C* . k expérimentale = k 0 .
log k expérimentale = log k 0 +
log
log g
i = - 0.5. z i2 . 
La courbe:
)D. Equation cinétique.
Le système étudié est la réduction lente de l' anion hexacyanoferrate (III) par l' acide ascorbique selon:
2 Fe(CN)63- + 2 H2O +
C6H8O6![]() 2 Fe(CN)64- + 2 H3O+ +
C6H6O6
2 Fe(CN)64- + 2 H3O+ +
C6H6O6
Réalisée dans les conditions stoechiométriques, de deux moles d' anion hexacyanoferrate (III) pour une mole d' acide ascorbique au départ, la réduction sera effectuée à force ionique VARIABLE par addition de nitrate de sodium NaNO3.
En abrégé la réduction s' écrira:
2 Fe III + AH2 + 2 H2O ![]() 2
Fe II + 2 H3O+ + A
2
Fe II + 2 H3O+ + A
On pose:
A t = 0:
(AH2) = a mol.L-1, (Fe III)
= 2a mol.L-1, (A) = (Fe II) = 0 mol.L-1.
A t = t:
(AH2) = a - x mol.L-1, (Fe III)
= 2a - 2x mol.L-1, (A) = x mol.L-1, (Fe II)
= 2x mol.L-1.
L' équation différentielle cinétique est la suivante:
 = 2.
k expérimentale . dt
= 2.
k expérimentale . dt
On intègre et on arrive à:
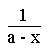 =
= ![]() + 2. k expérimentale. t
+ 2. k expérimentale. t
E = E° + 0.06.log 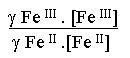
Les deux coefficients d' activité peuvent être
calculés grâce à la formule de DEBYE-HUCKEL.
A 25°C le potentiel normal E° du couple redox
étudié est égal à 0.358 V par rapport à
l' électrode normale à hydrogène.
La valeur mesurée par le millivoltmètre
est la différence de potentiel DE
entre le potentiel d' une électrode de mesure, à savoir ici
une électrode de platine, et le potentiel d' une électrode
au calomel, saturée en KCl.
On aura donc, si l' on exprime toutes les mesures en
millivolts:
E. Manipulation.
E.1. Solutions de départ.
E.1.a.
Dans une fiole de 200 mL introduire 10 mL d' acide ascorbique
à 1.5.10-2 mol.L-1.
Rajouter 10 mL d' acide nitrique à 0.2 mol.L-1.
Etendre à 200 mL avec de l' eau distillée.
E.1.b.
Introduire 25 mL de cette solution dans chacun des cinq flacons et les placer à 25°C.
E.1.c.
Dans les fioles de 25 mL introduire 5 mL de la solution
d' hexacyanoferrate (III) et un volume variable de solution de nitrate
de sodium à 0.15 mol.L-1.
Compléter avec de l' eau, agiter et laisser à
25°C.
Les volumes de solution de nitrate de sodium seront respectivement
de: 2, 5, 9, 14 et 20 mL.
E.2. Mesures.
Lorsque l' équilibre thermique est atteint faire le mélange des solutions directement dans la fiole thermostatée et commencer le relevé des mesures de différence de potentiel DE.
F. Résultats.
F.1.
Etablir la relation donnant la force ionique I en fonction
du volume V de solution de nitrate de sodium versé, ainsi que de
x, défini dans les équations différentielles
cinétiques plus haut.
F.2.
En supposant que I varie peu avec l' avancement de chaque
réaction, calculer sa valeur pour chaque expérience.
F.3.
Exprimer x en fonction de DE.
F.4.
Calculer les valeurs de k expérimentale correspondantes
pour chaque expérience.
F.5.
Sachant que les deux pKa de la viatmine C (fonction ène-diol)
sont respectivement 4.00 et 11.35 à 25°C, pourquoi faut-il rajouter
de l' acide nitrique, réputé être un acide fort?
F.6. Mécanisme de réaction.
Le mécanisme proposé est composé des trois étapes ci-dessous:
F.6.a. Première étape rapide: équilibre acidobasique:
AH2![]() AH-
+ H+
AH-
+ H+
[AH iiiiiiiiiiiiiiii Fe(CN)6]4-
Soit:
AH- + Fe(CN)63- ![]() [AH
iiiiiiiiiiiiiiii Fe(CN)6]4-
[AH
iiiiiiiiiiiiiiii Fe(CN)6]4-![]() AH
0
+
Fe(CN)64-
AH
0
+
Fe(CN)64-
F.6.c. Troisième étape: AH 0 peut, à son tour, réduire l' anion hexacyanoferrate (III) par la formation, puis la décomposition, d' un complexe activé:
[AH iiiiiiiiiiiii Fe(CN)6]3-
Soit:
AH 0 + Fe(CN)63- ![]() [AH
iiiiiiiiiiiii Fe(CN)6]3-
[AH
iiiiiiiiiiiii Fe(CN)6]3-![]() A
+ H+ + Fe(CN)64-
A
+ H+ + Fe(CN)64-
L' application de la théorie du complexe activé aux solutions ioniques mises en jeu ici permet de déterminer l' étape limitante.
Donner l' expression littérale de log k expérimentale
= f ( )
à l' aide des termes z A et z B et ce, dans
l' hypothèse de la formation intermédiaire du complexe activé
[AB*] z A + z B.
En déduire simplement que le terme k expérimentale
est fonction du produit z A . z B .
F.7. Interprétation des résultats.
F.7.1.
La vitesse de l' étape:
AH 0 + Fe(CN)63- ![]() [AH
iiiiiiiiiiiii Fe(CN)6]3-
[AH
iiiiiiiiiiiii Fe(CN)6]3-![]() A
+ H+ + Fe(CN)64-
A
+ H+ + Fe(CN)64-
n' est pas influencée par la force ionique I puisque
le produit z A. z B vaut 0 . ( - 3), soit 0.
Si cette étape est limitante alors on ne verra
pas de variation de k expérimentale avec I.
Si, au contraire, la force ionique I modifie la constante de vitesse k expérimentale, alors l' étape limitante est celle pour laquelle le produit z A . z B est non nul, à savoir:
AH- + Fe(CN)63- ![]() [AH
iiiiiiiiiiiiiiii Fe(CN)6]4-
[AH
iiiiiiiiiiiiiiii Fe(CN)6]4-![]() AH
0
+
Fe(CN)64-
AH
0
+
Fe(CN)64-
F.7.2.
A partir des résultats expérimentaux déduire lequel des deux transferts électroniques possibles est celui qui constitue l' étape lente.
Question subsidiaire si vous avez survécu:
Quelle est la signification de k 0?